Nous avions vu dans un précédent post que le génome de James Watson comptait un peu plus de 2 millions de variants par rapport au génome de référence; et qu'environ la moitié de ces variants étaient partagés avec Craig Venter.
Aujourd'hui, j'ai cherché à savoir si les densités des mutations à travers leurs génomes étaient semblables. Pour cela, j'ai fragmenté le génome en intervalles réguliers que j'appelle bins. J'ai ensuite compté pour chaque bin le nombre de variants chez Watson puis chez Venter. J'ai alors calculé la différence entre Watson et Venter pour chaque bin à l'aide d'un z-score.
Et voilà les résultats!
Pipeline
J'ai tout codé dans un pipeline disponible sur github.
Vous aurez besoin de Snakemake, de bedtools et du package R IdeoViz disponible depuis le site bioconductor.
Aucune donnée n'est nécessaire; tout se télécharge directement depuis le golden path d'UCSC. Vous pouvez d’ailleurs, si vous le voulez, lancer le pipeline sur d'autres génomes.
Exécution
La commande que j'utilise est la suivante :
snakemake -F --core 4 --config bin_size=100000 first=pgVenter second=pgWatson
- bin_size correspond à la taille des bins.
- first et second sont les noms des fichiers correspondant aux "personal genom (pg)" retrouvés dans UCSC.
- F sert à régénérer l'ensemble des fichiers dans le cas d'une seconde exécution.
- core spécifie le nombre de cœurs à utiliser.
Le schéma suivant représente les différentes étapes du pipeline.
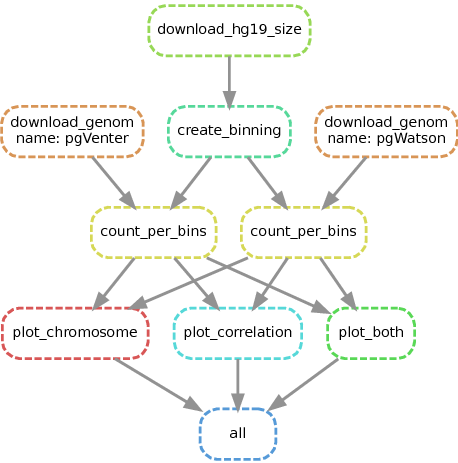
Graph du pipeline snakemake
Résultats
Si tout se passe bien vous devriez obtenir 3 images : correlation.png, both.png et ideogram.png que vous pouvez voir ci-dessous.
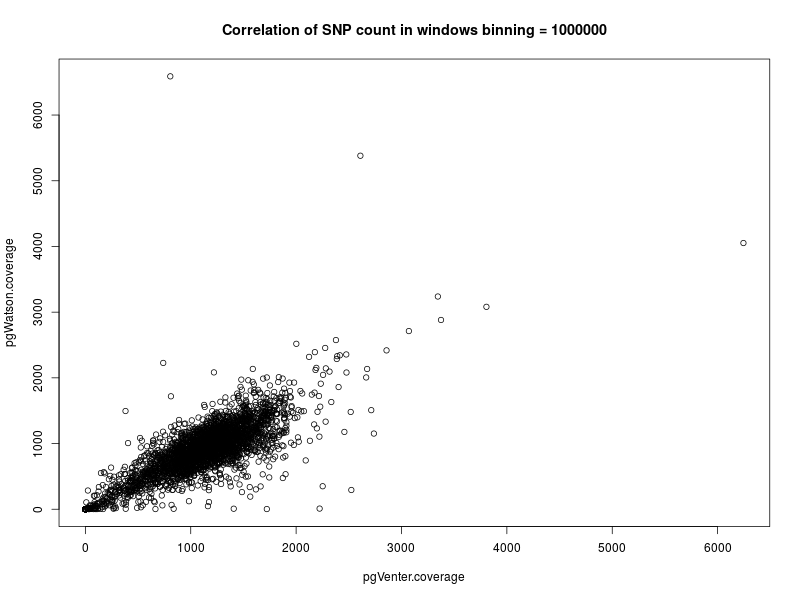
Corrélation du nombre de SNP par bin entre Watson et Venter avec des bins de 1 Mpb
Le graphique correlation.png représente le nombre de SNP par bin entre Watson et Venter en utilisant des bins d'1 million de paires de bases.
La corrélation est nette. Les régions riches en SNP chez Watson le sont également chez Venter.

Densités en variants par bin pour Watson (orange) et Venter (bleue)
Le graphique both.png montre les densités des variants par bin pour Watson (orange) et Venter (bleu). Les tendances générales des courbes semblent similaires.

Différence des SNPs par bin entre Watson et Venter sur tout le génome.
Le graphique ideogram.png montre les différences du nombre de SNP par bin entre Watson et Venter sur tout le génome. La différence est ici normalisée par un z-score. En zoomant, vous pouvez voir que les différences sont rarement significatives en restant inférieures à 2. En revanche, certaines régions notamment sur le bras long du chromosome X (avant dernier), montrent de grandes différences.
Conclusion
Les régions riches en mutations chez Watson le sont aussi chez Venter en utilisant des bins d'1 Mpb. Ceci peut s'expliquer par le contenu de la séquence. Il est possible que certaines régions soient plus susceptibles de muter à cause de leurs teneurs en non-codants, en zones répétées ou en autres choses. Il faudrait d'ailleurs que je regarde s'il y a une corrélation avec la teneur en exons, en GC...
La distribution dans le génome, fluctue quant à elle de la même façon chez Watson et Venter. On retrouve cependant quelques différences dans des zones précises. Peut-être des CNV...
Bref, prochain objectif, comparer ces courbes avec des données d'annotations style 1000 génomes et SNP !
Ce site est versionné sur GitHub. Vous pouvez corriger des erreurs en vous rendant à cette adresse
Go Top
comments powered by Disqus