Le microbiote et la métagénomique sont les deux mots tendances de ces dernières années dans les laboratoires de microbiologie. Derrière eux se cacherait les réponses à de nombreuses maladies comme le diabète, la maladie de Crohn et même l'autisme ou la schizophrénie.
Commençons donc par définir ces deux termes:
- Le microbiote est l'ensemble des micro-organismes (bactéries, virus, champignons, levures) vivants dans un environnement spécifique appelé microbiome. L'exemple typique est le microbiote intestinal. Votre intestin est composé de millions d'espèces bactériennes différentes formant une communauté écologique en symbiose avec votre organisme et nécessaire à son bon fonctionnement. Il joue entre autre un rôle de barrière vis-à-vis d'autres agents microbiens pathogènes. La destruction du microbiote intestinal par des antibiotiques est par exemple responsable des infections intestinales par Clostridium difficile.
Pour vous prouver l'importance du microbiome, retenez que le génome humain est composé d'environ 23 000 gènes. Le nombre de gènes retrouvés dans l'ensemble des micro-organismes du microbiome intestinal se compte en millions.
- La métagénomique est la méthode d'étude du microbiote. C'est une technique de séquençage et d'analyse de l'ADN contenu dans un milieu. A l'inverse de la génomique qui consiste à séquencer un unique génome, la métagénomique séquence les génomes de plusieurs individus d'espèces différentes dans un milieu donné. Une analyse typique de métagénomique vous donnera la composition d'un microbiome. C'est à dire quelles espèces sont présentes, leurs abondances et leurs diversités.
C'est en partie grâce à l’évolution majeure des technologies de séquençage haut débit et à la bioinformatique, que la métagénomique est aujourd'hui à notre portée.
Dans la suite de cet article, nous verrons uniquement la métagénomique bactérienne, plus particulièrement la métagénomique ciblé sur l'ARN 16S. Mais gardez bien en tête que les métagénomiques virales et fongiques, bien que plus rares, existent aussi.
Stratégie en métagénomique
Il existe deux grandes stratégies de séquençage en métagénomique : la stratégie globale et la stratégie ciblée.
- La métagénomique globale consiste à fragmenter tous les ADNs présents dans un échantillon en courts fragments et les séquencer à l'aide d'un séquenceur haut débit. D’où le nom de Shotgun sequencing. Les séquences (ou reads) obtenues sont ré-assemblées bioinformatiquement afin de reconstruire les génomes bactériens d'origine.
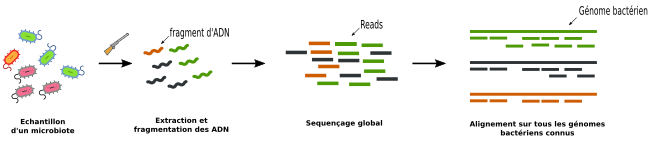
Stratégie globale : L'ensemble des ADNs présents dans un échantillon de microbiote sont séquencés.
- La métagénomique ciblée n'est pas de la métagénomique à proprement parler, mais de la métagénétique. Cette stratégie consiste à séquencer un unique gène au lieu d'un génome complet. Cependant le terme de métagénomique étant plus régulièrement employé pour décrire cette stratégie, je continuerai ainsi. Ce gène doit être commun à plusieurs espèces tout en présentant des régions suffisamment variables afin de discriminer une espèce. En bactériologie, le gène utilisé est celui de l'ARN 16S. Il s'agit d'un gène présent uniquement chez les bactéries.
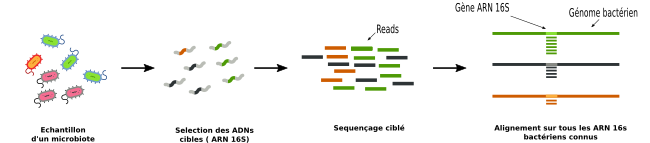
Stratégie ciblé : Seuls les ADNs du gène cible sont séquencés. En bactériologie, le gène cible est l'ARN 16S.
Chaque stratégie a son avantage. La métagénomique globale est plus précise dans le sens où elle séquence l'ensemble du génome d'une bactérie alors que la seconde ne s’intéresse qu'à un seul gène. Cette première stratégie permet par exemple de décrire le fonctionnement global du microbiote en séquençant l'ensemble des gènes présents.
La stratégie ciblée est quant à elle plus sélective. En effet, le gène de l'ARN 16S est présent uniquement chez les bactéries qui seules seront séquencées. La stratégie globale va séquencer tous les ADN présents dans le milieu sans discernement, qu'ils soient bactériens, viraux ou encore humains.
Enfin, les algorithmes de traitements des données issues d'un séquençage ciblé sont beaucoup plus simples que les assemblages de génomes nécessaires dans le séquençage global. Pour comprendre cette complexité, essayez de mélanger toutes les pièces de 200 puzzles différents et tentez de retrouver les modèles originaux. C'est la problématique de la métagénomique globale.
On ne s’intéressera ici qu'à la stratégie 16S, utilisée en bactériologie. C'est un bon point de départ pour commencer !
L' ARN 16S
Vous connaissez les ribosomes ? Ces petits organelles dans la cellule formés de deux sous-unités permettant la traduction de l'ARN en protéine. Et bien chez la bactérie, et uniquement chez elle, la petite sous unité est formée de l'ARN 16S.

Structure secondaire de l'ARN 16S avec ses différentes boucles.
Il s'agit d'un ARN non codant composé d'environ 1500 nucléotides possédant des régions constantes et variables. Il suffit d'aligner la séquence d'ARN 16S de différentes espèces bactériennes pour s'en rendre compte. Comme vous pouvez le voir sur la figure ci-dessous, certaines régions sont constantes entre les bactéries alors que d'autres régions sont variables.
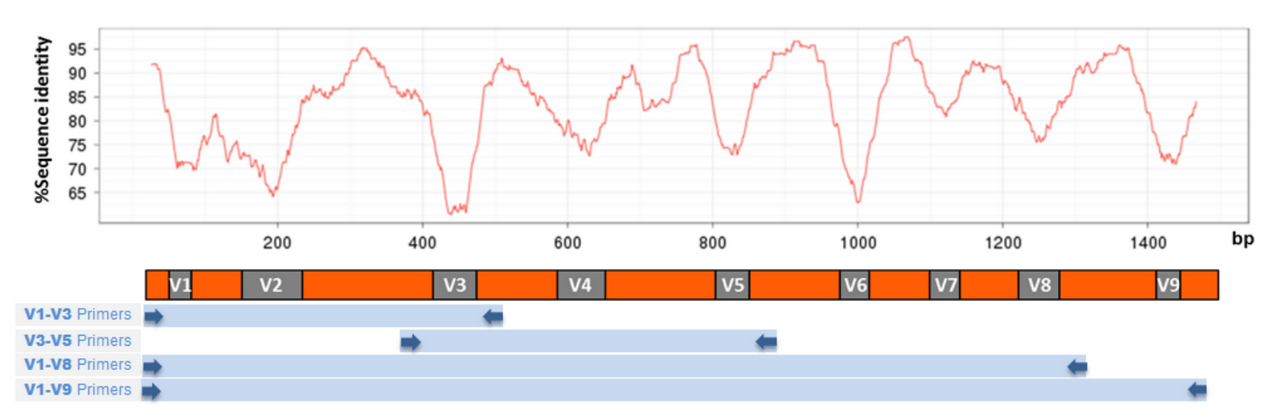
Similarités des séquences d'ARN 16S entre plusieurs bactéries. Sous le graphique figurent les différents couples d'amorces utilisables.
Les régions variables n'ont pas de rôle fonctionnel important et peuvent diverger au cours de l’évolution sous l'effet des mutations neutres.
C'est ce qui va nous permettre de discriminer les taxons bactériens au sein du microbiome. A chaque taxon correspondra une séquence particulière au niveau des régions variables. Il s'agit de la signature du taxon.
Les régions constantes vont permettre quant à elles de capturer l'ensemble des ARN 16S. Ces régions étant identiques chez toutes les bactéries, il est possible de construire des amorces comme pour une PCR afin de sélectionner la région d’intérêt.
En réalité, seule une partie de l'ARN 16S est séquencée car les séquenceurs haut débit ne peuvent pas séquencer d'un coup les 1500 nucléotides de l'ARN 16S (enfin... sauf le Pacbio). Le couple d'amorce V3-V5, que vous pouvez voir sur la figure 3, permet par exemple de séquencer une région de 500 nucléotides contenant 3 régions variables.
Assignent taxonomique
Une fois le séquençage réalisé, c'est au tour des bioinformaticiens de prendre le relais. Un fichier contenant l'ensemble des reads (séquences) est obtenu après séquençage. Après plusieurs étapes de filtrage et de nettoyage de ces données, il faut assigner à chaque séquence le nom de la bactérie. Pour cela, deux stratégies existent.
- La stratégie close-reference consiste à comparer chaque séquence aux séquences présentes dans une base de donnéees avec un seuil en général de 97% de similarité. Greengene, Silva et RDP sont les bases de données d'ARN 16S les plus connues. Cette stratégie a le mérite d'être rapide mais son principal problème est d'ignorer les séquences absentes des bases de données. Pour palier à ce problème, la deuxième stratégie peut être utilisée.
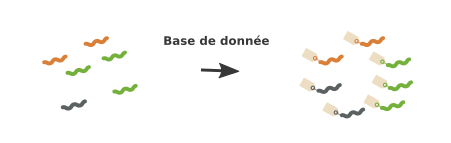
Stratégie 1. Chaque séquence est recherchée dans une base de données et assignée à son taxon.
- La stratégie appelée de novo, n'utilise pas de base données mais consiste à comparer les séquences entre elles puis les regrouper par similarité. Les clusters ainsi formés élisent une séquence consensus qui peut à son tour être annotée par une base de données ou rester comme telle définissant alors une espèce inconnue.
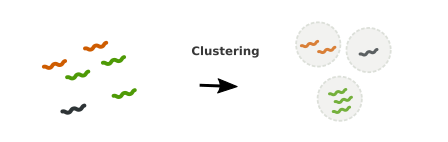
Stratégie 2. Les séquences sont comparées entre elles pour former des groupes similaires ou clusters.
Une fois l’assignation taxonomique réalisée, il suffit de compter le nombre d'espèces présentes dans chaque échantillon et de construire la table des OTUs.
La table des OTUs
Le point de départ de toutes analyses en métagénomique est la construction de la table des OTUs (operationnal taxonomic unit). La notion d'espèce est difficile avec les bactéries, on parle plutôt d'OTU pour définir un ensemble de bactéries similaires à plus de 97 %.
La table des OTUs est un tableau à double entrées contenant le nombre de séquences par OTU et par échantillon. On parle d'abondance. Ces abondances absolues sont normalisées afin de rendre les échantillons comparables. Plusieurs méthodes de normalisation existent, mais la plus courante est d'utiliser les pourcentages. Sur la figure ci-dessous, les échantillons 1 et 3 ont tous les deux une abondance absolue de 3 en bactéries rouges. En pourcentage, leurs abondances relatives deviennent 42,8 % et 75 % respectivement.
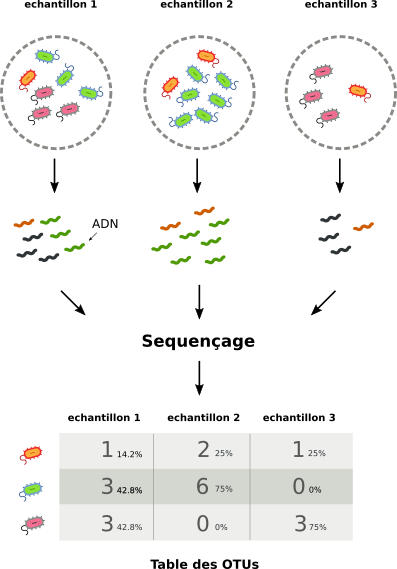
Tables des OTUs obtenues à partir de plusieurs échantillons
Analyse des données
Diversité Alpha
La diversité alpha est une mesure indiquant la diversité d'un échantillon unique. Le nombre d'espèce est par exemple un indicateur d'alpha diversité.
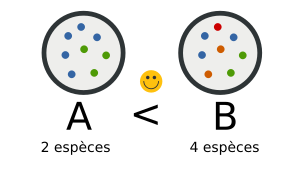
B est plus diversifié que A car il contient deux fois plus d'espèces
Mais comme vous pouvez le voir dans la figure ci-dessous, Le nombre d'espèce n'est pas toujours adapté. C'est pour cette raison que d'autres indicateurs existent.
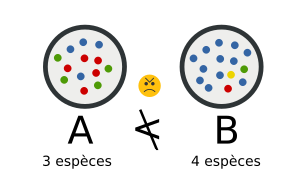
B contient plus d'espèce mais semble moins diversifié
L'indice de Shannon ou entropie de Shannon est un exemple d'alpha diversité répondant à ce problème. Cette indice reflète aussi bien le nombre d'espèce que leurs abondances. Sa formule est la suivante :

Indice de Shannon. Pour chaque espèce faire la somme des fréquences multiplié par le log des fréquences
La figure A précédente contient 13 espèces, dont 4 vertes, 5 rouges et 4 bleues. La diversité de shannon pour A est donc :
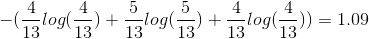
En faisant de même pour B, on retrouve alors une diversité plus faible de 0.72.
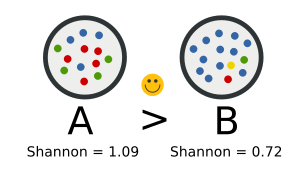
L'entropie de A est supérieur à celle de B
Les autres indicateurs répondent chacun à des problèmes différents. L'indice Chao1 estime le nombre d'espèce réel dans l'environnement à partir du nombre d'espèce dans l'échantillon. Il y a aussi l'indice de Simpson, de Fisher et l'indice ACE. Faite un tour sur ce site pour avoir plus des informations plus détaillées.
Le graphique ci-dessous montre les différences de diversité alpha du microbiote intestinal en fonction du régime alimentaire.
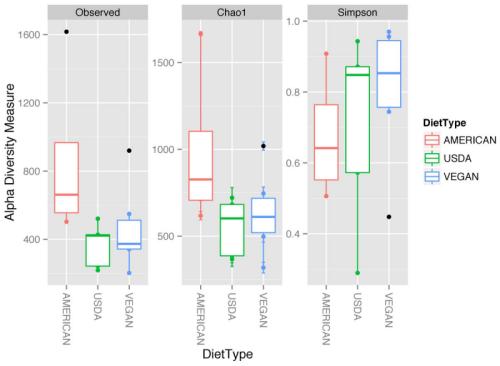
Diversité alpha du microbiote intestinal en fonction du régime alimentaire.
Source
Source
Diversité Beta
La diversité bêta consiste à mesurer la diversité des espèces entre les échantillons. On procède le plus souvent à l'analyse multivariée de la matrice des OTUs en ayant recours aux méthodes d'ordinations comme l'analyse en composantes principales. Pour faire simple, imaginons que notre table des OTUs soit composée de 2 bactéries et 6 échantillons. La représentation sur un graphique serait facile en utilisant 2 axes (1 par bactérie). Chaque point de ce graphique serait un échantillon dont les coordonnées représentent l'abondance pour chaque bactérie. La figure de gauche ci-dessous illustre cet exemple. En colorant ces points sur une variable attachée aux échantillons, comme le site de prélèvement, on pourrait découvrir des groupes distincts, comme l'illustre la figure de droite.
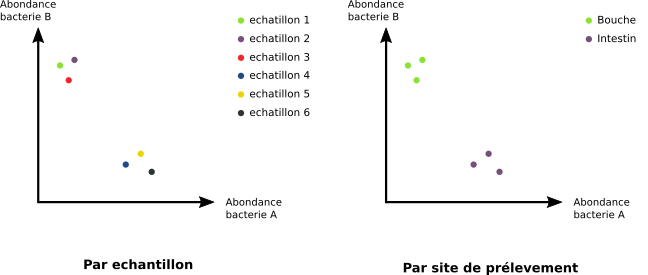
Chaque point représente un échantillon réparti sur les deux axes en fonction de leurs abondances. Certains échantillons semblent associées entre eux.
Bien entendu, il y a plus de deux bactéries différentes dans un microbiome. Ce qui nécessite un nombre d'axe impossible à représenter graphiquement. Les méthodes d'ordination répondent à ce problème en projetant la variabilité de tous ces axes sur 2 axes pouvant être visualisés.
L'analyse en composantes principales (ACP) est un exemple d'ordination. Il en existe bien évidemment d'autres. La plus couramment utilisée en métagénomique est une jumelle de l'ACP, l'analyse en coordonnées principales (PCoA) que je ne détaillerai pas.
Une fois la représentation réalisée, on cherche alors des groupes de points et la variable explicative que l'on visualise à l'aide d'une couleur. Sur la figure ci-dessous, l'analyse de plusieurs échantillons provenant de différents sites anatomiques révèle les compositions propres à chaque site.
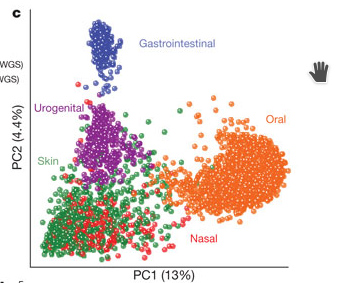
Analyse en composantes principales de différents échantillons microbiens provenant de différents sites anatomiques.
Source
Source
Conclusion
La métagénomique est un sujet complexe en plein essor qui nécessite une connaissance précise des différentes techniques pour éviter tout écueil. De nombreux biais peuvent intervenir à toutes les étapes, tant du coté biologique que bioinformatique. D'ailleurs, l'assignation taxonomique que je décris dans cet article reste simple et naïve. D'autres méthodes plus complexes mais valables statistiquement sont préférables. Par exemple la méthode dite de « Minimum Entropy Decomposition » permet de classer les OTU en s'abstenant du seuil théorique des 97 %.
Enfin, si vous voulez approfondir la métagénomique, je vous invite très fortement à regarder les vidéos de Dan Knights (un dieu en métagénomique) disponibles sur YouTube!
Références
- Cours en vidéo de Dan Knights
- Génomique et métagénomique bactérienne: applications cliniques et importance médicale
- Enterotype of the human gut microbiome
- Structure, function and diversity of the healthy human microbiome
- Outil : QIIME
- Outil : Vsearch
- Outil :Philoseq
Remerciements
@Thibaud_GS
@Piplopp
@pausrrls
@Oodnadatta
Ce site est versionné sur GitHub. Vous pouvez corriger des erreurs en vous rendant à cette adresse
Go Top
comments powered by Disqus