Vous connaissez le jeu du chaos? Il s'agit d'une construction géométrique très simple permettant de faire apparaitre des fractales. La construction la plus connue est le triangle de Sierpinski que vous pouvez dessiner vous-même avec un papier et un crayon:
- Dessiner un triangle en numérotant les trois sommets A,B,C.
- Puis dessiner dedans un point P choisi au hasard .
- Tirer alors un nombre aléatoire correspondant à A,B ou C.
- Si par exemple vous tirez le A, dessiner le point correspondant au milieu du segment [PA].
- Ce nouveau point appelez le P, puis répéter la procédure de façon itérative en partant du nouveau point.
Si tout se passe bien, et avec beaucoup de temps, vous devrez voir apparaitre le triangle de Sierpinski.
Le jeu du chaos appliqué à l'ADN
Au lieu d'utiliser un dé, nous pouvons utiliser une séquence d'ADN pour choisir les sommets avec cette fois un carré ou chaque sommet correspond aux nucléotides A,C,G,T. Pour chaque base lue dans la séquence, dessiner le point correspondant au centre du segment [P-nucléotide] puis continuer comme vu précédement jusqu'au dernier nucléotide.
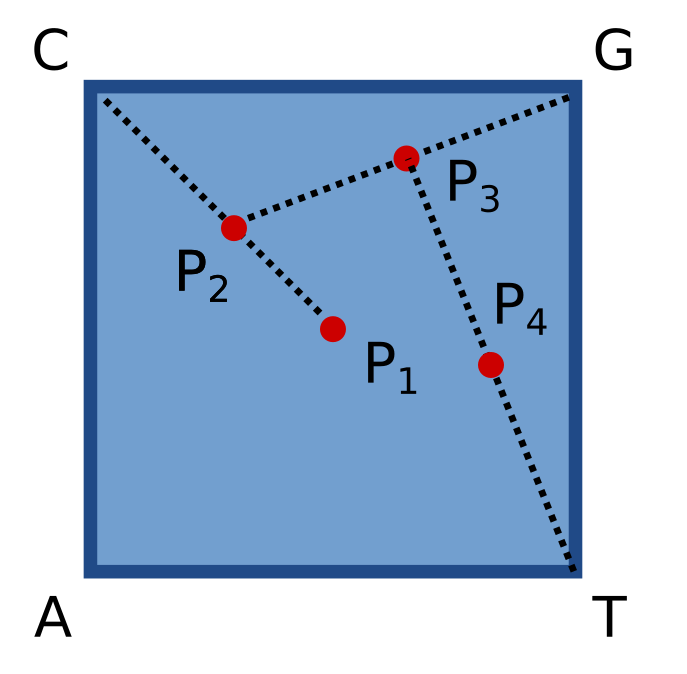
Construction d'une CGR pour la séquence CGT. A partir du centre, trouver le point P2 centre du segment [P1-C]. Puis P3 centre du segment [P2-G] et enfin P4 centre de [P3-T].
Avec un programme informatique c'est plus rapide. On peut alors executer l'algorithme sur de très longues séquences comme des génomes entiers. Et ça donne de très jolies images:

Exemple de CGR obtenu à partir de plusieurs espèces. source
Pour comprendre ces graphiques appelés CGR (Chaos Game Representation), garder à l'esprit qu'à chaque point correspond une partie de la séquence lue. Par exemple il y a un point correspondant aux 4 premiers nucléotides et un autre point correspondant aux 20 premiers nucléotides. Si vous réfléchissez un peu, vous devinerez que toutes séquences commençant par un A dessine un point dans le quart inférieur gauche, celles commençant par un G dans le quart supérieur droit, ainsi de suite. Mais nous pouvons aller encore plus loin. Toutes les séquences commençant par CG, se trouvent dans le quart supérieur gauche du quart droit. Toutes les séquences commençant par TAG, dans le quart inférieur droit, du quart inférieur gauche, du quart supérieur droit. Cette dichotomie illustrée sur la figure ci-dessous permet d'associer à chaque séquence une coordonnée unique. Et si vous n'avez pas compris, allez faire un tour sur cette page et tapper n'importe quel séquence dans la barre de recherche.
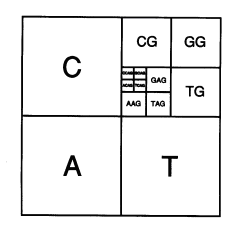
Dichotomie de la CGR. Par exemple, toutes les séquences commencant par TAG se trouve dans une zone précise.
Une méthode pour compresser l'ADN
À part être jolie, à quoi ça sert ? Et bien plusieurs choses. Cette représentation apporte une information globale (sur toute la séquence) et une information locale (sur le contenu de la séquence). Par exemple, sur la figure suivante, vous pouvez voir un "trou" dans le quart G (supérieure droite). Ce motif se répète à plusieurs échelles ( dans les sous-quarts) et correspond à une dispersion des répétitions CG. ( Ce pattern serait observé uniquement chez les vertébrés ).

CGR d'une région contenant le gène de la beta globuline sur le chromosome 11. source
On peut s'en servir aussi comme une signature. Ou encore pour visualiser des réarrangements...
Mais ce que je préfère c'est l'utilisation de cet algorithme pour compresser une séquence d'ADN. En effet, plus haut je vous ai dit qu'à chaque séquence il y a un unique point. Par exemple avec la séquence ACGT, les coordonnées du dernier point sont unique à la séquence. Il n'y a que la séquence ACGT qui permet de produire ce point. On peut donc représenter n'importe quelle séquence par un couple de coordonnées (x,y) !
Avec l'algorithme que nous venons de voir et nos ordinateurs actuels nous pouvons compresser 32 nucléotides en utilisant un couple de nombres à virgule (x,y). C'est pas mal, mais il y a mieux. Un article récent montre qu'il est possible de compresser 1024 nucléotides avec un couple d'entiers (x,y) en modifiant la méthode de calcul. Au lieu de calculer le milieu d'un segment, la somme entre les deux points est calculé en utilisant une puissance de 2 dans l'équation.
N'importe quelle séquence de moins de 1024 nucléotides peut ainsi être écrite en utilisant 3 nombres : la longueur de la séquence, et les coordonnées x, y.
On pourrait alors très bien imaginer un algorithme, qui découpe une très longue séquence d'ADN en bloc de 1024 nucléotides et compresse chaque morceau avec l'ensemble mis bout à bout. Génial non ?
Ceci est une séquence de 3072 (1024*3) nucléotides écrit sur une ligne!!!
(52332,12313)(5744,14)(1242,75575)
Source
- lifenscence
- Chaos game representation of gene structure.(Jeffrey, H. J. 1990).
- Additive methods for genomic signatures
- Encoding DNA sequences by integer chaos game representation
Remerciements
Merci à @Natir pour cette découverte
Ce site est versionné sur GitHub. Vous pouvez corriger des erreurs en vous rendant à cette adresse
Go Top
comments powered by Disqus