Lors de la mitose, l'ADN des cellules eucaryotes s'organise en chromosomes condensés et bien délimités. C'est l'image que nous avons tous d'un chromosome. Mais le reste du temps, pendant l'interphase, ces chromosomes ressemblent davantage à une boule de spaghetti emmêlée dans tous les sens : ce que l'on appelle la chromatine.
Aujourd'hui, les technologies de Capture de Conformation des Chromosomes nous révèlent l'organisation spatiale de cette chromatine, qui en réalité semble loin de l'anarchie mais plutôt organisée de façon fonctionnelle. C'est ce que nous allons découvrir tout de suite avec les TADs (Topologically Associated Domain).
Analyser l'organisation spatiale de la chromatine
Il existe toute une famille de technologies permettant d'évaluer l'organisation spatiale des chromosomes au moment de l'interphase. C'est la Capture de Conformation des chromosomes (Chromosom Conformation Capture). Cette méthode se décline sous plusieurs formes que vous trouverez sous le nom de (3C,4C,5C,HiC,ChiA-PET). Elles reposent toutes sur le même principe qui est d'identifier sur la chromatine, des régions en contact physique. Imaginez la chromatine comme un long ruban d'ADN, formant plein de boucles en se repliant sur elle-même. Par ces technologies, vous allez pouvoir savoir qu'une région x de ce ruban est en contact physique avec une autre région y.
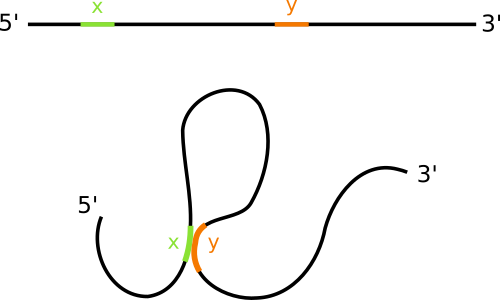
Figure 1. Haut. Vision linéaire du génome. Bas. Vision spatiale du génome et identification d'une zone de contact (orange)
Comment ça fonctionne ?
L'idée générale consiste à capturer les deux régions d'ADN en contact (x et y) et construire un fragment d'ADN hybride contenant le fragment x à une extrémité et le fragment y de l'autre (Figure 2). Ce fragment hybride est alors identifié par différents techniques de biologie moléculaire.
Tout d'abord, les régions de contact sont figées en créant des liaisons covalentes grâce à du formaldéhyde. C'est l'étape du cross-linking. L'ADN est ensuite digéré avec des enzymes de restriction pour ne garder que les régions de contact. Puis on réalise une ligation des extrémités du cross-link pour obtenir des fragments d'ADN hybrides.
Ces fragments peuvent alors être identifiés par les différentes méthodes de biologie moléculaire. Par exemple, la méthode 3C est une simple PCR tandis que la méthode Hi-C est un séquençage haut débit de l' ensemble de fragments hybrides obtenus à partir d'un génome. C'est cette dernière que je vais détailler.
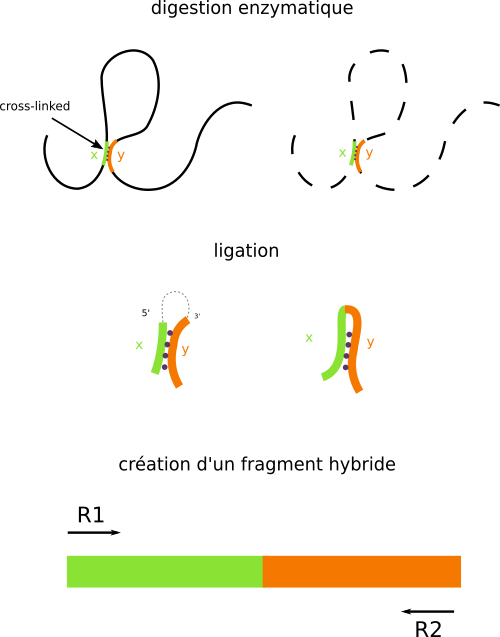
Figure 2. Fixation des régions de contact avec du formaldéhyde (cross-linking) puis digestion de l'ADN avec une enzyme de restriction. Grâce à une ligase, les deux extrémités du cross-link sont reliées. Après un reverse cross-linking, le fragment hybride est obtenu. Celui-ci va pouvoir être séquencé en paired-end sur de l'Illumina. Le read R1 correspondra à la région x et le read R2 à la région y.
Méthode Hi-C
À partir de la méthode décrite au-dessus, on va pouvoir créer une libraire de séquençage, c'est-à-dire générer un ensemble de fragments hybrides correspondant à l'ensemble des zones de contact de la chromatine. Cette librairie est lue sur un séquenceur Illumina qui a la particularité de faire du séquençage en paire (paired-end), c'est-à-dire pouvoir lire un fragment d'ADN dans les deux sens. Pour chaque fragment d'ADN lu, nous obtenons ainsi une paire de reads R1 et R2 qui correspondent aux deux régions de contact x et y (Figure 2). On aligne ces reads sur le génome de référence afin de leur attribuer des coordonnées génomiques. Connaissant les paires de reads, nous pouvons enfin savoir si une région x est en contact avec une région y.
Par exemple, si dans nos données, il existe un read R1 s'alignant sur le gène A et un read R2 s'alignant sur le gène B, nous pouvons "conclure" qu'il y a interaction entre le gène A et le gène B.
Visualiser les données Hi-C
Pour représenter l'ensemble des régions de contact provenant d'une expérience Hi-C, on utilise une carte de chaleur (heatmap). Cette carte est une matrice n x n montrant le nombre d'interactions entre deux positions données du même chromosome. La technologie ne permettant pas d'avoir une résolution à la base exacte, les positions sont des intervalles de taille fixe.
La valeur de chaque cellule est le nombre de paires de reads entre deux positions données. Plus la couleur d'une cellule est rouge, plus il y a d'interaction entre les deux positions correspondant à cette cellule.
La figure 3 gauche, montre comment construire une heatmap pour un chromosome. La figure 3 droite, montre une heatmap sur des données réelles Hi-C pour le chromosome 14. La diagonale rouge vif signifie que les régions très proches dans la séquence, sont en contact physique, ce qui semble logique. Notons par ailleurs que la matrice est symétrique. En effet, "x" interagit avec "y", est équivalent à "y" interagit avec "x". Pour cette raison, on préfère représenter les données Hi-C par une demi heatmap, ce qui nous donne un triangle (figure 4).
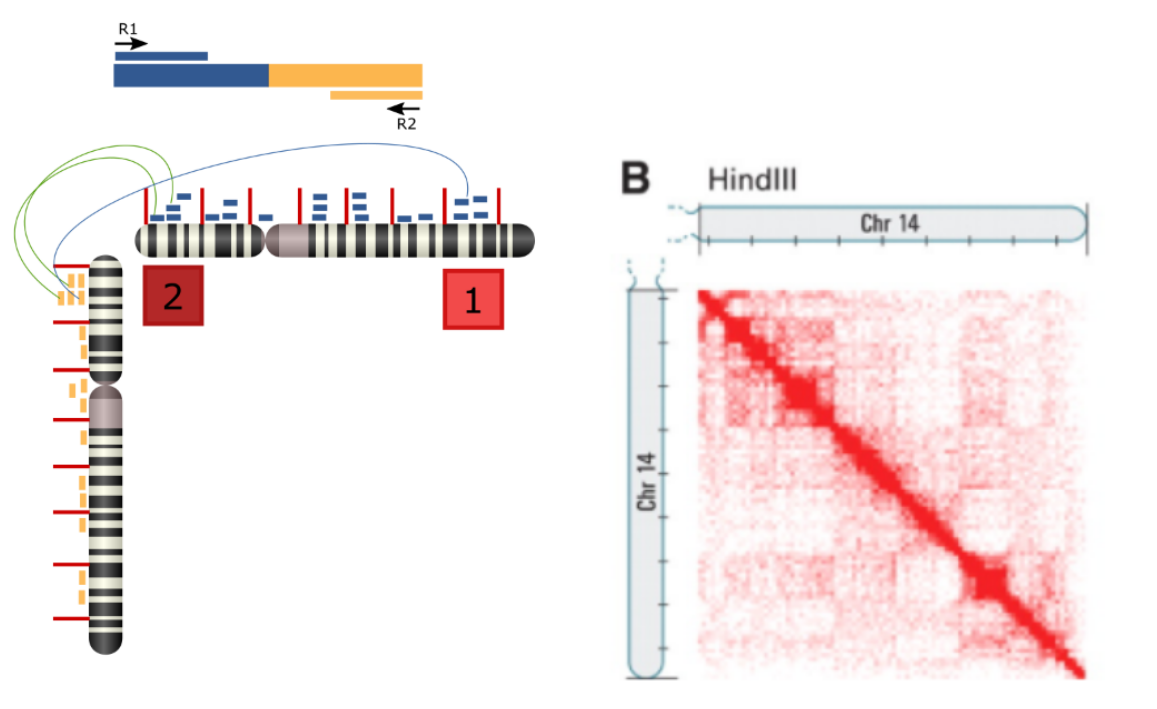
Figure 3. Gauche : le chromosome est découpé en intervalles de taille fixe. Après alignement, chaque read est associé à un intervalle. On comptabilise alors le nombre de paires existantes pour deux intervalles donnés. Sur la figure de gauche, il y a 2 paires entre les deux extrémités "p" et une paire entre l'extrémité "p" et "q". Droite : données réelles Hi-C sur le chromosome 14. Notez la symétrie de la matrice autour de la diagonale ainsi qu'une allure en damier. source
Les TADs
En observant la heatmap de plus près (Figure 4), vous distinguerez des triangles rouges d'allure fractale qui ressortent clairement. Ces triangles correspondent à un ensemble de régions qui interagissent toutes ensemble mais qui sont isolées du reste. Ces domaines, ce sont nos fameux TAD. Imaginez-les comme des boules de noeud sur notre ruban D'ADN. Chaque noeud contient des régions qui interagissent avec les autres régions de ce même noeud mais jamais avec d'autres. Pour vous faire une idée, deux TADs sont magnifiquement illustrés par mes soins en bas de la figure 4.
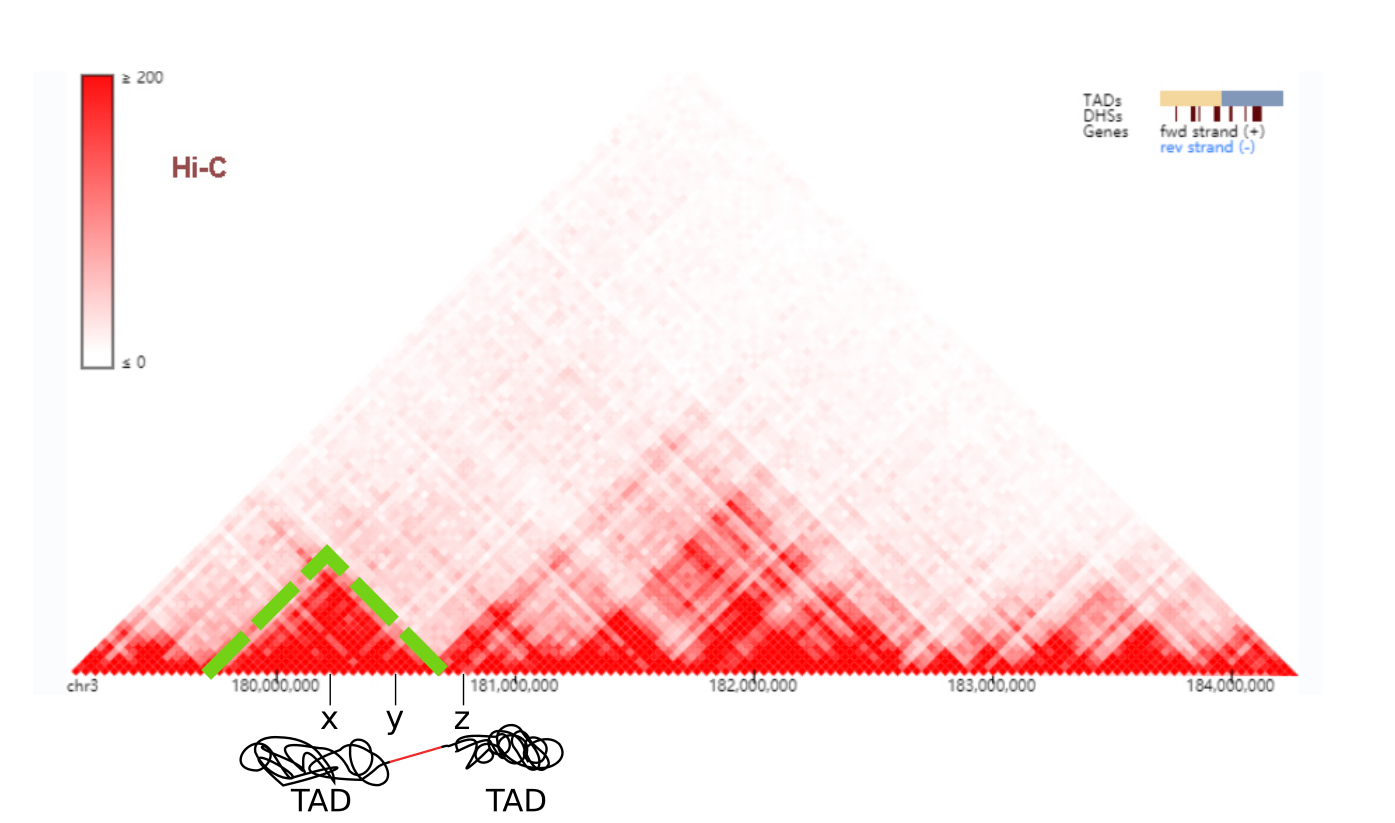
Figure 4. Visualisation d'une région du chromosome 3. Les TADs sont des domaines qui interagissent et sont observés ici par des triangles rouges. Sur cette figure, la région x,y et z sont à egale distance les unes des autres. Cependant x et y appartiennent au même TAD tandis que z appartient à un autre différent. source
Fonction des TADs
Aujourd'hui, la fonction des TAD n'est pas totalement élucidée. Mais il est clair qu'ils jouent un rôle important dans la régulation de l'expression des gènes. Nous savons depuis longtemps que les gènes sont régulés par des séquences promotrices situées en amont des gènes. Mais il existe aussi des régions très éloignées du gène qui peuvent moduler la transcription. Ce sont les amplificateurs (enhancers) et les inactivateurs (silencers) qui respectivement activent ou répriment la transcription. Par exemple, en repliant l'ADN dans l'espace, l'enhancer et le promoteur vont pouvoir interagir et moduler la transcription (Figure 5).
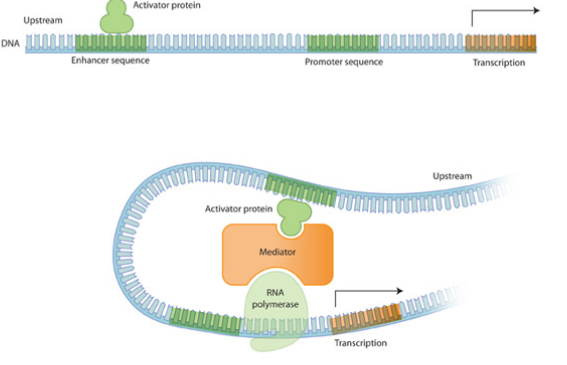
Figure 5. Schéma de la régulation de la transcription via les amplificateurs. En se répliant, l'ADN met en contact l'amplificateur et le promoteur d'un gène.
Il est alors évident que pour agir, un amplificateur doit se situer dans le même TAD que ses gènes cibles. Plusieurs gènes au sein du même TAD peuvent ainsi être co-régulés par le même amplificateur.
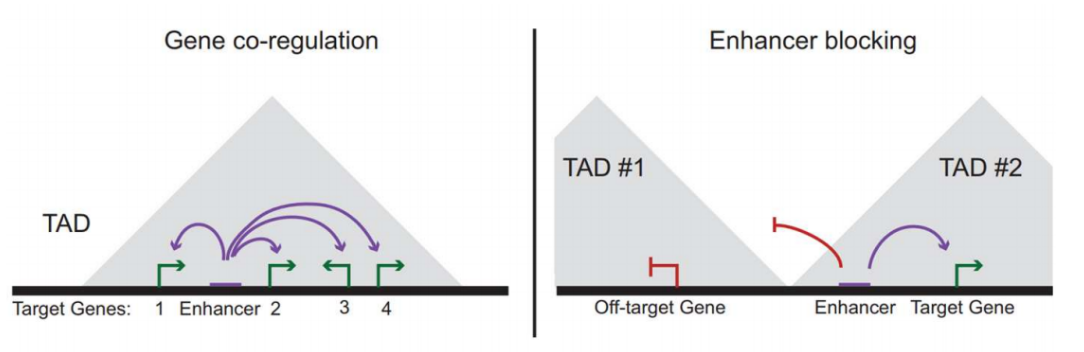
Figure 6. Un amplificateur peut interagir avec les gènes de son TAD mais pas avec un autre.
Une autre région importante dans la régulation est l'isolateur (insulator) qui se situe entre deux TAD en empêchant leur fusion. Une étude a par exemple montré qu'une délétion dans un isolateur est responsable de la fusion de deux TADs en un seul. Les deux noeuds bien distincts ne forment plus qu'un seul gros noeuds. Les enhancers du premier TAD sont alors capables d'interagir avec un gène du deuxième TAD, entraînant une sur-expression délétère pour l'organisme.
Formation des TADs
La formation des TADs a été récemment mise en évidence en validant le modèle de Loop extrusion. Ce mécanisme fait intervenir la cohésine et les protéines CTCF qui reconnaissent des motifs autour des TADs, et font glisser la chromatine au travers d'anneaux. Les deux vidéos suivantes vous montrent clairement la formation de ces structures.
Simulation du modèle Loop extrusion
Visualisation de Loop extrusion temps-réel
Conclusion
La découverte d'une organisation spatiale de la chromatine a changé notre vision du génome. Les chromosomes étaient le support rigide de l'information génétique. Ils sont maintenant les acteurs d'une régulation fine contrôlé par l'épigénétique. L'exploration dans ce domaine nous permettra de mieux comprendre le fonctionnement du génome dans son intégralité, et justifiera certainement le séquençage complet des patients atteints de maladies génétiques.
Références
- La thèse d'un collègue
- Analysis methods for studying the 3D architecture of the genome
- Topological domains in mammalian genomes identified by analysis of chromatin interactions
- bioinformaticsinstitute
- bioinfo-fr
- Outil: TAD disease
- Outil: Hi-C Browser
Ce site est versionné sur GitHub. Vous pouvez corriger des erreurs en vous rendant à cette adresse
Go Top
comments powered by Disqus