La pandémie mondiale de Covid-19 a créé un élan sans précédent dans la production scientifique de données. Notamment, les données sur les génomes du virus produites par séquençage haut débit qui permettent aujourd'hui d'identifier de nouvelles mutations comme la N501Y du variant anglais B.1.1.7 où une Asparagine (N) est remplacée par une Tyrosine (Y) à la position 501 de la protéine S.
Le génome du SARS-CoV-2 étant tout petit (30 kbases) par rapport au génome humain (3 Gbases), l'analyse bioinformatique peut se faire avec un ordinateur personnel. Cette analyse est donc l'occasion d'un très bon exercice pour se familiariser avec les outils d'alignement et de détection de variants.
Dans ce billet, je vous propose de suivre pas-à-pas l'analyse d'un génome de SARS-CoV-2 à partir des données brutes générées par un séquenceur Illumina. Pour cela, j'ai récupéré les données de séquençage de 245 échantillons provenant d'un laboratoire de l'état du Delaware aux États-Unis. Plus précisément une paire de fichiers Fastq par échantillon contenant les courtes séquences, appelées reads, lus par le séquenceur. Je les ai ensuite toutes alignées sur le génome de référence de Wuhan pour en extraire les mutations que j'ai annotées. Enfin, j'ai reconstruit les génomes de chaque échantillon afin de pouvoir attribuer le nom de leurs lignées.
Pour réaliser cette analyse, il vous faudra de préférence un terminal Linux avec différents outils que vous pouvez installer avec conda. Sinon un notebook est à votre disposition à cette adresse.
Analyse de l'échantillon SRR13182925
Source des données
Je connais 2 sources principales pour récupérer des données de séquençage. SRA du NCBI et la base européenne ENA. J'ai choisi cette dernière par convenance.
En fouillant, j'ai d'abord découvert le projet ICOG UK constamment mis à jour et disposant de pas moins de 206 000 génomes à l'heure où j'écris ce billet.
Mais pour mon petit PC de bureau, c'est trop de données. Nous nous contenterons pour le moment des 245 génomes du projet PRJNA673096 produit par le Delaware Public Health Lab aux États-Unis. Il s'agit de données produites sur un Illumina MiSeq en Amplicon.
Téléchargement des données avec EnaBrowserTools
Nous pouvons télécharger les fichiers Fastq directement depuis le site de l'ENA. Mais il existe un outil python en ligne de commande, enaBrowserTools, qui va nous faciliter la tâche. Pour l'installer, il suffit de suivre la documentation:
-
enaDataGet nous permet de télécharger les données associées à un échantillon.
-
enaGroupGet nous permet de télécharger l'ensemble des données d'un projet.
Pour la suite de ce billet, nous nous contenterons d'analyser uniquement l'échantillon SRR13182925. Pour télécharger ses fichiers Fastq associés, taper la commande suivante:
enaDataGet -f fastq SRR13182925
Cette commande crée un dossier SRR13182925 contenant 2 fichiers Fastq :
- SRR13182925_1.fastq.gz
- SRR13182925_2.fastq.gz
Ces 2 fichiers contiennent les courtes séquences lues, appelées reads, d'environ 150 bases provenant du génome viral trouvé dans l'échantillon. Ces reads sont lus dans les deux sens par le séquenceur. Pour cette raison, il y a 2 fichiers, un par sens de lecture.
Vous pourrez en avoir un aperçu à l'aide la commande suivante :
zcat SRR13182925_1.fastq.gz|awk 'NR % 4 == 2 {print $0}'
AGATAATACAGTTGAATTGGCAGGCACTTCTGTTGCATTACCAGCTTGTAGACGTACTGTGGCAGCTAAACTACCAAGTAC
ATATTGGCTTCCGGTGTAACTGTTATTGCCTGACCAGTACCAGTGTGTGTAC
TCGTAACAATCAAAGTACTTATCAACAACTTCAACTACAAATAGTAGTTGTCTGATATCACACATTGTTGGTAGATTATAACGATAGTAGTCATAATCGCTGATAGCAGCATTACCATCCTGAGCAAAGAAGAAGTGTTTTAATTCAACAGAACTTCCTTCCTTAAAGAAACCCTTAGACACAGCAAAGTCATAGAAGTCTTTGTTAAAATTACCGGGTTTGACAGTTTGAAAAGCAACATTGTTAGTAAGTGCAGCTACTGAAAAGCACGTAGTGCGT
ACATTACACATAAACGAACTTATGGATTTGTTTATGAGAATCTTCACAATTGGAACTGTAACTTTGAAGCAAGGTGAAATCAAGGATGCTACTCCTTCAGATTTTGTTCGCGCTACTGCAACGATACCGATACAAGCC
GTTAGATAGCACTCTAGTGTCAAATCTACAAACAATGGAATTAGCAGGATATCTATCGACATTGCAATTCCAAAATAGGCATACACCATCTGTGAATTTGTCAGAATGTGTGGCATAAGAATAGAAT
GTTTATTACCCTGACAAAGTTTTCAGATCCTCAGTTTTACATTCAACTCAGGACTTGTTCTTACCTTTCTTTTCCAATGTTACTTGGTTCCATGCTATACATG
etc ...
Pour plus de précision sur le format Fastq, lisez la spécification du format.
Alignement des reads sur le génome de Wuhan
L'alignement consiste à aligner les reads présents dans les fichiers Fastq (150 bases) sur un génome de référence du Sars-CoV-2 qui fait lui environ 30 000 bases.
Nous devons d'abord télécharger ce génome de référence au doux nom de NC_045512.2. Il s'agit d'un fichier Fasta que vous pouvez récupérer depuis le site ou via la commande suivante :
wget -O wuhan.fasta https://www.ncbi.nlm.nih.gov/sviewer/viewer.cgi?tool=portal&save=file&log$=seqview&db=nuccore&report=fasta&id=1798174254&extrafeat=null&conwithfeat=on&hide-cdd=on
Avant de procéder à l'alignement avec l'outil bwa, il est nécessaire d'indexer ce génome. On fera de même avec l'outil samtools qui nous servira par la suite :
bwa index wuhan.fasta
samtools faidx wuhan.fasta
L'alignement est réalisé à l'aide de la commande ci-dessous qui nous créera un nouveau fichier SRR13182925.sam contenant les reads associés à leur position d'alignement sur le génome:
bwa mem wuhan.fasta SRR13182925_1.fastq.gz SRR13182925_2.fastq.gz > SRR13182925.sam
Ce fichier SAM est un fichier texte. On lui préfère sa version binaire, le BAM plus légère et indexable. Faisant d'une pierre deux coups, je trie le fichier par position, le convertit au format BAM et l'indexe avec les commandes ci-dessous:
samtools sort -O bam SRR13182925.sam > SRR13182925.bam
samtools index SRR13182925.bam
Visualisation de l'alignement
Pour visualiser cet alignement, vous pouvez utiliser le logiciel IGV disponible à cette adresse. Une fois lancé, chargez d'abord le génome de Wuhan depuis le menu Genomes > Load Genome From Server en cherchant SARS-Cov-2. Puis chargez le fichier SRR13182925.bam précédemment créé via File > Load From File. Vous obtiendrez ainsi la vue suivante où j'ai zoomé sur le gène S pour visualiser une mutation.
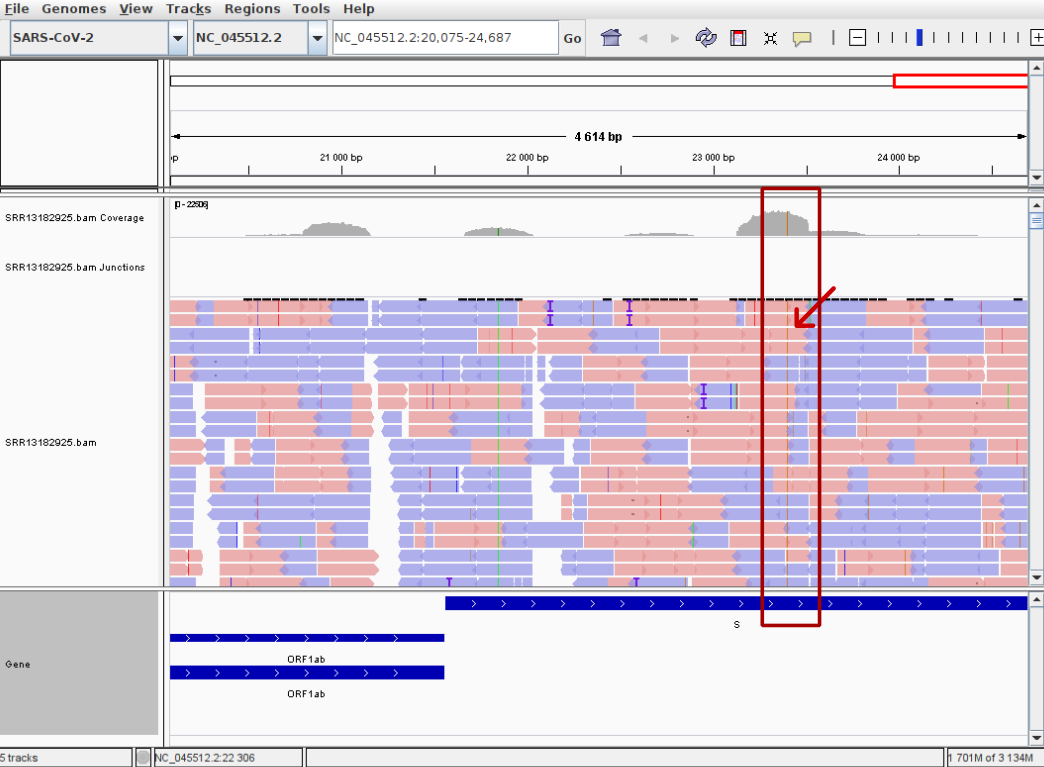
Visualisation des reads alignés sur le génome de référence avec le logiciel IGV. La flèche montre une mutation située sur le gène S visible sur l'ensemble des reads
Appel des variants et annotation
Vous pourriez parcourir l'alignement visuellement et chercher toutes les mutations. Mais il est préférable de procéder de façon automatique grâce à un variant caller. Pour cela j'utilise freebayes, qui à partir du fichier BAM, crée un fichier VCF contenant l'ensemble des variants détectés. Avec SnpSift, on garde uniquement les variants de bonne qualité avec un score superieur à 30 et on compresse avec bgzip le fichier pour pouvoir l'indexer avec tabix:
freebayes -f wuhan.fasta -p1 -C10 SRR13182925.bam|SnpSift filter "QUAL > 30" - > SRR13182925.vcf
bgzip SRR13182925.vcf
tabix -p vcf SRR13182925.vcf.gz
Le fichier VCF obtenu contient uniquement les positions et les bases mutées. Pour avoir plus d'information, j'annote ce fichier avec SnpEff qui me donnera entre autres le nom de la mutation en nomenclature HGVS ainsi que le gène où il se situe:
snpEff -Xmx10G -v NC_045512.2 SRR13182925.vcf.gz > SRR13182925.ann.vcf
Il me suffit maintenant d'extraire de ce fichier les informations pertinantes. Pour cela, j'utilise SnpSift filter associé à SnpSift extracFields pour afficher les mutations dans un tableau à deux colonnes avec le nom de la mutation et le nom du gène ou elle se situe:
SnpSift filter "(exists ANN[0].HGVS_P)" > SRR13182925.ann.vcf
|SnpSift extractFields - "ANN[0].HGVS_P" "ANN[0].GENE"
ANN[0].HGVS_P ANN[0].GENE
p.Phe924Phe ORF1ab
p.Thr1665Thr ORF1ab
p.Pro4715Leu ORF1ab
p.Thr95Asn S
p.Asp614Gly S
p.Ser68Phe E
p.Asp72Asp E
Vous pouvez également ouvrir le fichier SRR13182925.ann.vcf via l'interface graphique de Cutevariant. C'est un logiciel de mon cru en version bêta! Donc, soyez indulgent.
Création du génome consensus
Pour reconstruire la séquence du génome de l'échantillon à partir du fichier VCF, nous pouvons utiliser bcftools qui nous génère un fichier Fasta:
bcftools consensus SRR13182910.vcf.gz -f genome/whuan.fasta --sample unknown > SRR13182910.fa
Nous pouvons alors utiliser l'outil pangolin pour asigner le nom de la ligné à ce génome. La nomenclature est défini dans ce papier:
pangolin SRR13182910.fa
Nous obtenons alors l'identifiant B.1.1.119 dont vous pouvez trouver la déscription sur cette page. Et comme nous pouvions s'y attendre, elle est trouvé en grande partie chez des nord-américains.
Analyse des 245 génomes
Distribution des variants
J'ai d'abord téléchargés tous les fichiers Fastq du projet via la commande suivante :
enaGroupGet -f fastq PRJNA673096
Puis j'ai réalisé un pipeline avec Snakemake disponible ici. Il reprend les mêmes étapes vues plus haut à la seule différence que l'ensemble des variants détectés est colligé dans un même fichier VCF.
Après quelques heures de calcul, j'ai finalement obtenu ce fichier VCF qui m'a permis d'analyser la fréquence des variants le long du génome que j'ai reporté dans le graphique suivant.
Au total, j'ai trouvé environ 630 variants répartis le long du génome dont 4 mutations particulièrement fréquentes.
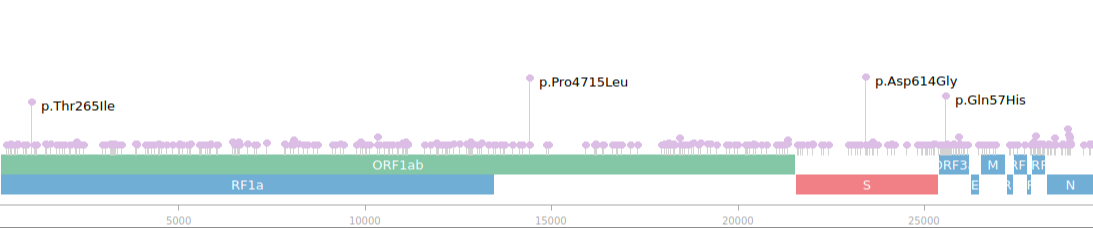
Repartition des variants trouvés parmi les 245 génomes avec leurs fréquences
Ces 4 mutations sont probablement la conséquence d'un processus de sélection.
En googlant, je trouve ce papier présentant les mutations Thr265Ile and Gln57His comme exclusif à la population Nord-Américaines. Ce qui colle bien avec l'origine de nos données.
Le variant Pro4715Leu est également dominant et affecte la polymérase RNA-dependent (RdRp) catalysant la réplication de l'ARN. Peut-être que cette mutation modifie la fidélité de la réplication impactant ainsi son taux de mutation et donc son evolvabilité, c'est-à-dire sa capacité à évoluer.
Plus intéressant, est le variant Asp614Gly situé sur le gène (S) de la protéine Spike, là même ou se situe le variant anglais en permettant au virus de pénétrer plus facilement les cellules humaines. Cette mutation Asp614Gly serait apparue en Europe début 2020 et serait maintenant la forme majoritaire (source).
Distribution des lignées
J'ai également reconstruit chaque génome que j'ai assigné à sa ligné avec l'outil pangolin. J'obtient la distribution suivant:
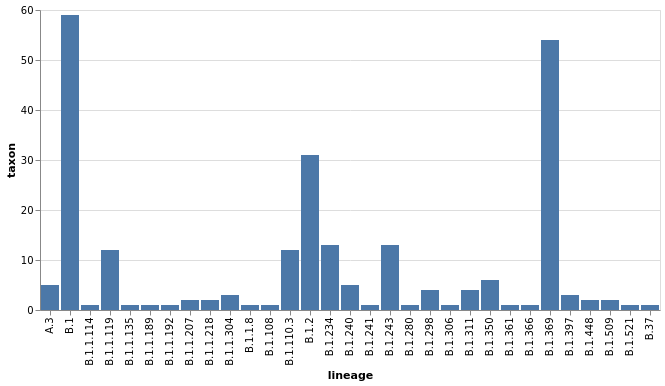
Distribution des lignées de virus
Nous pouvons voir ici la présence de ligné B.1.2 en grand nombre. D'après le site cov-lineages, elle est exclusivement Nord-américaine est descend de la ligné B.1 apparu précocement et que l'on trouve partout dans le monde.
Quand à la ligné B.1.369, elle a d'abord été vu en Océanie puis aux Etats-Unis à partir de Juin 2020.
Bref, tout qui colle avec l'origine nord-américaine de mes données!
Conclusion
Un très bon exercice rapide qui en intéressera certainement plus d'un. En tout cas, je me suis bien amusé ce week-end à apprendre un tas de chose interessante sur ce maudit virus.
N'hésitez donc pas à reprendre mon pipeline si vous en avez besoin.
J'aimerai maintenant apprendre à reconstruire un arbre phylogénétique à la fois dans le temps et dans l'espace. Ce qu'ils ont fait sur le site nextrain, est vraiment trop stylé! Cela permet de tracer l'évolution de ce virus qui, on ne le rappellera jamais assez, est expliquée par la théorie de l'évolution de Darwin.
Ce site est versionné sur GitHub. Vous pouvez corriger des erreurs en vous rendant à cette adresse
Go Top
comments powered by Disqus