Le séquençage de nouvelle génération (NGS: Next Generation Sequencing) est la révolution biotechnologique de ces dernières années, en permettant de séquencer de grandes quantités d'ADN en des temps records.
À titre d'exemple, le projet human genome a coûté 3 milliards de dollars sur 13 ans entre 1990 et 2003 pour séquencer le génome humain en utilisant des séquenceurs de type Sanger répartis dans plusieurs laboratoires à travers le monde. Aujourd'hui, avec un séquenceur NGS Illumina HiSeq X, en trois jours, on peut séquencer trois génomes humains pour 1000 dollars chacun. Le graphique ci-dessous, que vous verrez régulièrement, montre l'évolution du coût de séquençage par million de nucléotides au cours du temps. Et encore, ce graphique s'arrête en 2015. La société Illumina a déjà promis le génome à $100 d'ici deux ans avec le nouveau séquenceur Illumina NovaSeq.
Cet article est un avant-gout très vulgarisé pour découvrir les bases du séquençage haut débit.
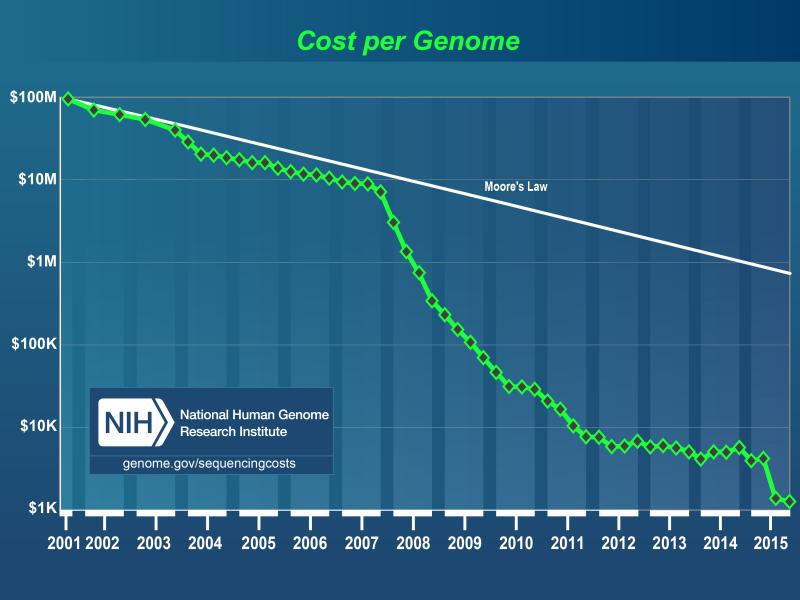
Diminutions du coût du séquençage par nucléotides au cours des dernières années
Un séquençage à haut-débit
Imaginez que votre génome s'assimile à un gros livre de plus de 3 milliards de caractères (nucléotides) écrit avec les lettres A,C,G et T. Séquencer, c'est lire le contenu de ce livre. Vous pouvez soit le lire entièrement, c'est-à-dire séquencer l'ensemble de votre génome. Soit lire certaines pages ou chapitre, c'est-à-dire faire du séquençage ciblé. Les séquenceurs actuels ne peuvent lire que des courts fragments d'ADN qu'il faut ensuite assembler pour reconstruire le texte d'origine. Par exemple, les séquenceurs de premières générations de type Sanger sont capables de lire des fragments d'environ 800 caractères en 1 heure sur 1 capillaire. Si vous faites le calcul, vous verrez rapidement que pour atteindre les 3 milliards de nucléotides, il vous faudra plus d'une vie pour réussir à séquencer votre génome (>400 ans). Pour aller plus vite, l'idée est de lire plusieurs fragments en même temps, c'est-à-dire paralléliser le séquençage. Les plus performants des séquenceurs Sanger, peuvent paralléliser jusqu'à 96 fois en utilisant 96 capillaires. On a donc 96 x 800 nucléotides lus en 1 heure. C'est pas encore ça. Les séquenceurs NGS de deuxièmes générations sont capables, eux, de lire des fragments de 150 à 300 pb mais jusqu'à 20 milliards de fragments à la fois!!!
Librairie de séquençage
Ce qu'on appelle une librairie, est l'ensemble des fragments d'ADN que l'on veut séquencer. Pour créer une librairie, deux méthodes sont à retenir si l'on veut séquencer l'ensemble du génome ou des régions d'intérêts.
Méthode globale
Lancez le livre en l'air et tirez dessus au shotgun pour faire une pluie de fragments d'ADN aléatoire. C'est ce qu'on appelle stricto sensu, la stratégie shotgun. Cette méthode est utilisée par exemple pour séquencer des génomes entiers.
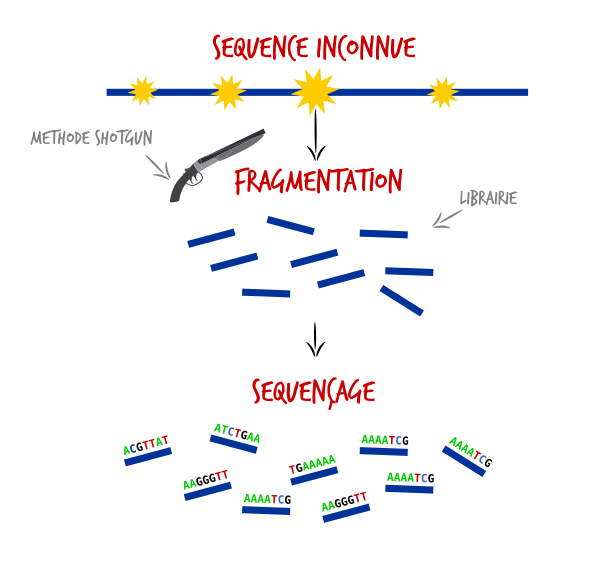
La stratégie shotgun consiste à fragmenter l'ADN en séquence aléatoire puis à les séquencer.
Plusieurs méthodes existent pour fragmenter l'ADN:
- Fragmentation par sonication : En envoyant des ultra-sons à la bonne fréquence, on casse l'ADN en morceaux de tailles précises.
- Fragmentation enzymatique : L'utilisation d'enzymes de restriction permet de couper l'ADN au niveau de certains motifs.
Méthode ciblée
On ne veut pas forcément lire l'ensemble du génome. On peut vouloir par exemple séquencer uniquement la partie codante (exome), qui je le rappelle, représente moins de 2% ; ou simplement séquencer une liste de gènes (panel de gènes) associée à une maladie.
Dans tous les cas, il faut enrichir la librairie en sélectionnant uniquement les fragments d'ADN désirés. Deux techniques sont à retenir:
- L'enrichissement par capture : Après fragmentation, les fragments d'ADN sont filtrés en s'hybridant à des séquences complémentaires disposées sur une plaque ou en milieu liquide. Les fragments d'ADN qui ne s'hybrident pas sont éliminés.
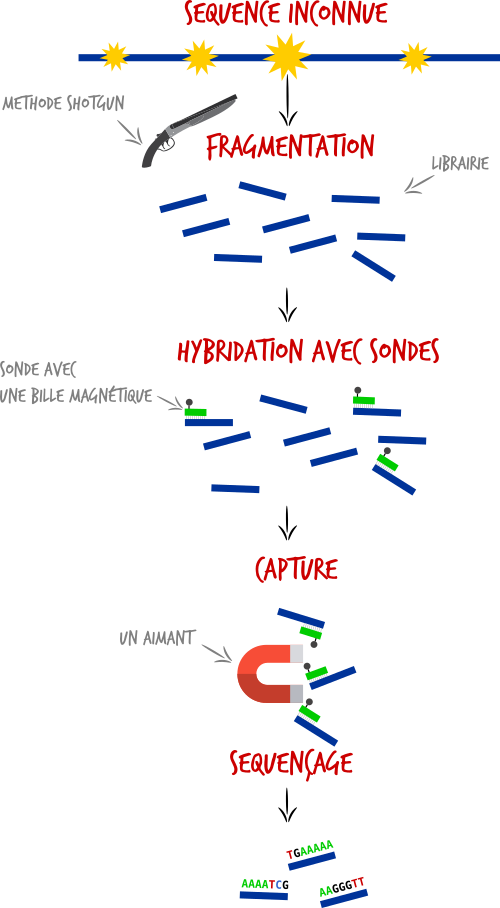
Exemple d'enrichissement en phase liquide grâce à des billes magnétiques
- Enrichissement par PCR : Les fragments désirés sont amplifiés par PCR. Nous pouvons amplifier une seule région avec un couple d'amorces (simplex), ou alors amplifier plusieurs régions avec plusieurs couples d'amorces (multiplex). Dans cette stratégie, toutes les séquences d'un même amplicon seront identiques.
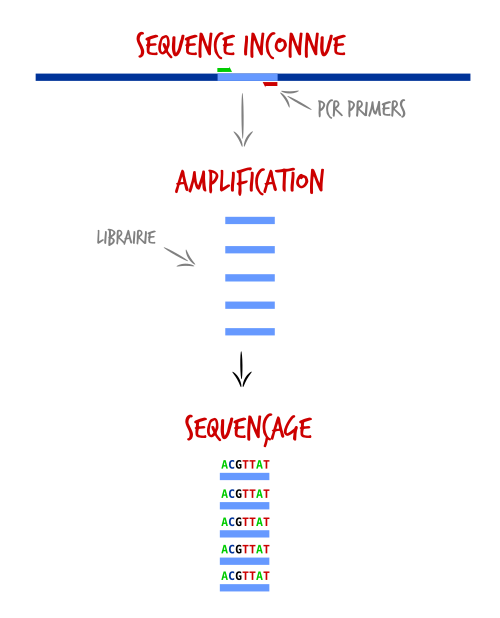
La stratégie par amplicon produit des séquences identiques
Sequençage
Il existe différentes méthodes de séquençage:
- Le séquençage par synthèse (Illumina).
- Le pyroséquençage (Roche 454).
- La ligation (SOLid Thermofisher).
- La détection des ions H+ (Proton Thermofisher).
Dans l'ensemble, le principe général reste le même. Chaque fragment est d'abord cloné plusieurs fois afin d'amplifier le signal. Puis le brin complémentaire de chaque fragment cloné est synthétisé. À chaque incorporation d'un nucléotide, un signal est détecté. De la lumière pour Illumina ou une variation de pH sur du Proton. À la fin du séquençage, chaque fragment a été séquencé en parallèle. L'ensemble des données est enregistré dans un fichier Fastq.
Exemple de séquençage sur Proton
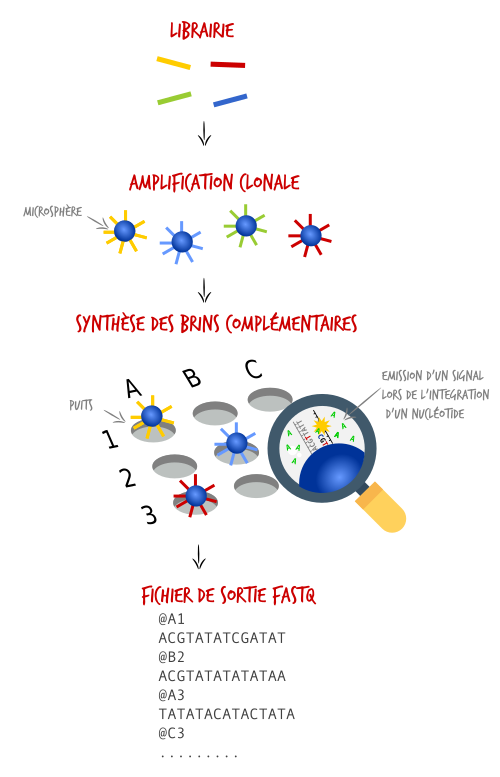
Schéma simplifié d'un séquençage type Proton
Pour plus de détail sur les techniques de séquençage, jettez un oeil sur les belles vidéos commerciales ci-dessous:
Alignement des séquences
À la fin du séquençage, la chimie fait place à la bioinformatique. Les séquences des fragments, qu'on appelle maintenant des "reads", sont sauvegardées dans un fichier Fastq contenant les séquences et leurs scores de qualité (score Phred). Ce score évalue la confiance du séquençage. Par exemple le séquenceur vous dira que pour tel reads, la probablité que le quatrième nucléotide soit un 'A' est de 99,9%. Ce score appelé 'Q score' est encodé en caractère ASCII pour chaque nucléotide d'un read.
Télécharger ici un exemple pour voir à quoi ça ressemble.
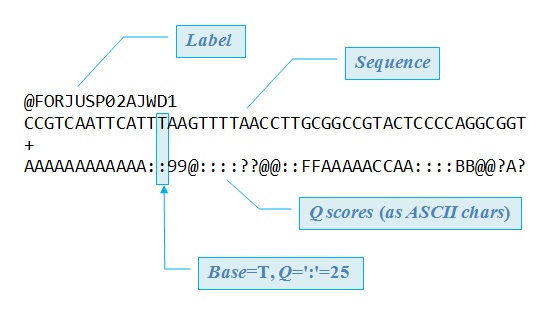
Aperçu d'un read dans un fichier fastq. Le score de qualité associe à chaque nucléotide un caractère ASCII
Mais le travail est loin d'être fini. Ce que nous avons, ce sont uniquement des courtes séquences de 150 pb en général. Ce que nous voulons, c'est obtenir la séquence complète d'un gène ou d'un génome entier. Pour cela, il faut reconstruire un puzzle en réalisant un alignement. Deux méthodes existent :
- Assemblage de novo : Il s'agit de résoudre un puzzle sans son modèle. Les fragments d'ADN qui sont chevauchants permettent petit à petit de reconstruire ce qu'on appelle un contig. L'assemblage des contigs entre eux permet d'obtenir un scaffold. Cette technique est très couteuse en termes de calcul. Des algorithmes bioinformatiques comme les graphe de Bruijn, permettent de résoudre ce problème. Cette méthode est principalement employée pour reconstruire des génomes non connus.
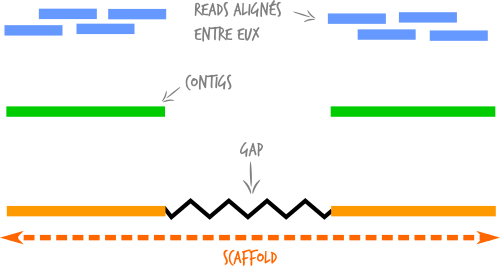
L'alignement de novo consiste à aligner les reads entre eux
- Alignement avec référence: Il s'agit toujours de résoudre un puzzle. Mais cette fois, en s'aidant d'un modèle. Par exemple, une version du génome humain (hg19).
Chaque read est aligné sur cette référence. La complexité de calcul est plus simple qu'avec l'alignement de novo. On utilise en général l'algorithme de Burrows Wheeler permettant de rechercher de manière efficace une correspondance entre les reads et la référence. Après cet alignement, on obtient un fichier BAM associant à chaque reads ses coordonnées génomiques. C'est à dire le chromosome et la position.
On appelle la profondeur, le nombre moyen de reads qui se superpose et recouvrement, l'étalement des reads sur la zone d'intérêt.

L'alignement avec référence consiste à aligner les reads sur une référence
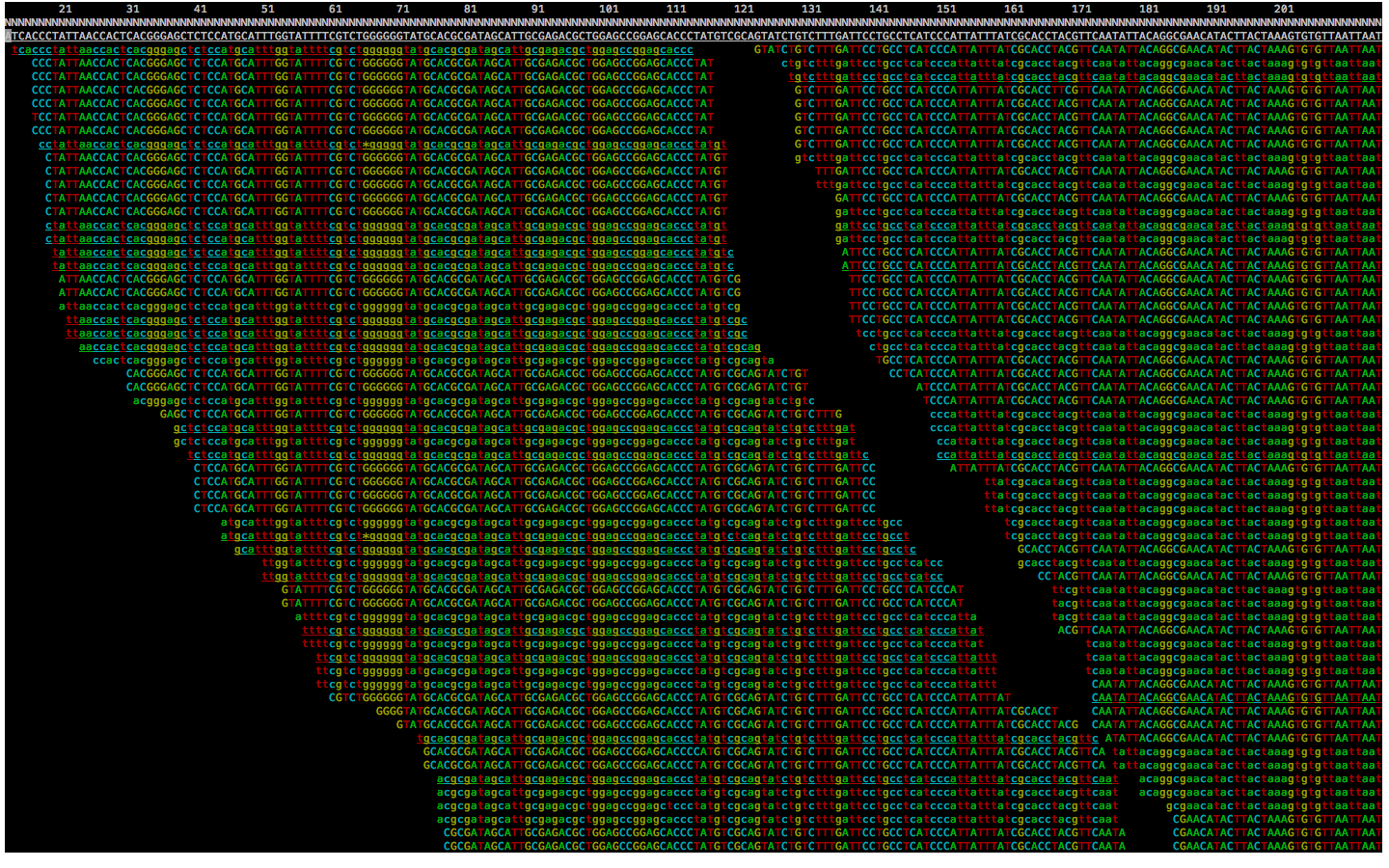
Visualisation d'un alignement réel avec Samtools
Évaluation d'un séquenceur
Avec tout ça, vous êtes capable d'évaluer la capacité d'un séquenceur comme vous le feriez avant l'achat d'un PC ou d'une voiture.
Les capacités d'un séquenceur sont définies par :
- La longueur des reads produits (L)
- Le nombre de reads produits (n)
- Le nombre de nucléotides lu: (L x n)
- Le temps de séquençage
- La qualité du séquençage
Allez faire un tour sur le site d'Illumina pour comparer les modèles entre eux.
Pour finir, les séquenceurs de 3ème génération
À peine sortie, ces technologies sont déjà devancées par les séquenceurs de 3ème générations. Ce sont des séquenceurs capables de générer de très longs reads sans avoir besoin de cloner les fragments pour amplifier le signal. C'est pour cette raison qu'on les appelle aussi "Single molecule sequencing". En revanche, ces nouvelles techniques produisent encore beaucoup d'erreurs de séquençage. Les deux leaders de ce Next Next Generation Sequencing sont Nanopore et PacBio Science qui termine une guerre de brevet.
La miniaturisation de ces séquenceurs sera peut être un jour disponible chez tout bon médecin généraliste qui vous diagnostiquera votre prédisposition d'infarctus ou d'Alzheimer en quelques heures. Effrayant ou rassurant, à vous de choisir!
vous ne rêvez pas... C'est le SmidgIon d'Ofxord Nanopore, un séquenceur qui se branche sur un IPhone
Références
Remerciements
Ce site est versionné sur GitHub. Vous pouvez corriger des erreurs en vous rendant à cette adresse
Go Top
comments powered by Disqus