Un nombre important de maladies génétiques sont causées par des mutations ponctuelles, c'est-à-dire des mutations à l'échelle du nucléotide. Les mécanismes sous-jacents, que vous devez certainement connaître, sont la substitution, la délétion ou l'insertion de nucléotides dans la séquence d'un gène. Les mutations dynamiques sont peut-être moins connues mais elles sont pourtant responsables de plusieurs maladies génétiques héréditaires. Notamment une maladie neurodégénérative appelée la maladie de Huntigton.
Définition
La maladie de Huntigton est une maladie génétique autosomique dominante touchant environ 1 personne sur 10 000. Elle est causée par une anomalie du gène HTT (anciennement IT15) situé sur le chromosome 4 et codant pour la protéine Huntigtine. La mutation d'un seul des deux allèles suffit pour provoquer la maladie. Un parent a donc une chance sur deux de la transmettre.
Cliniquement, la maladie s'accompagne d'une altération de la fonction intellectuelle et du comportement. Les mouvements anormaux sont très caractéristiques. Ce sont des mouvements brusques incontrôlés de la face et des membres qu'on appelle chorée de Huntigton (vidéo ci-dessous).
La maladie survient généralement entre 30 et 50 ans, mais comme nous allons le voir, peut survenir plus tôt en fonction de la mutation.
Mutation dynamique
Le mécanisme mutationnel de la maladie de Huntigton est lié à l'expansion d'un motif de nucléotides (CAG) dans la partie codante de l'exon 1 du gène. Nous avons déjà discuté de ces répétitions dans l'article sur les empreintes génétiques. Le codon CAG code pour l'acide aminé glutamine. Son expansion se traduit donc par une augmentation d'homopolymère de glutamine dans la protéine qui lui confère un pouvoir toxique dans le cerveau. Plus précisément l'atteinte se situe au niveau des neurones GABAergiques du striatum. Chez les sujets sains, la taille de la répétition varie entre 6 et 35. Entre 41 et 180, elle est pathologique. La zone intermédiaire entre 35 et 41 définit une zone floue, parfois symptomatique, parfois asymptomatique, qu'on appelle zone de pénétrance réduite.
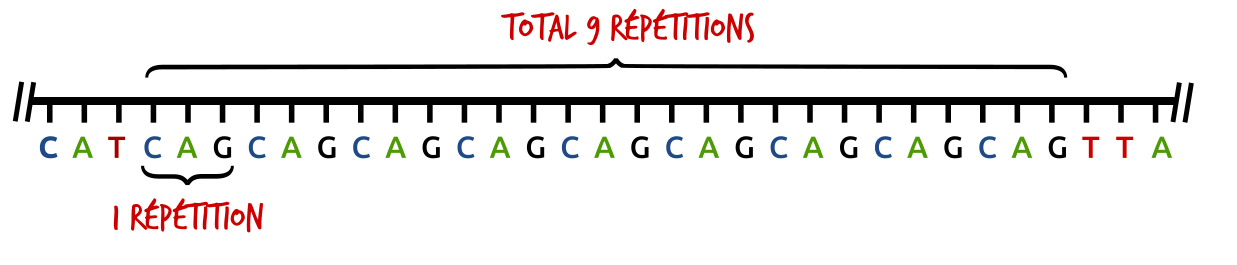
Expansion des triplets CAG dans la maladie de Huntigton. Sur cette figure, il y a 9 répétitions
Phénomène d'anticipation
Plusieurs études ont montré que la longueur de la répétition est corrélée à l'âge où débute la maladie. Les longues répétitions sont responsables des formes juvéniles qui peuvent apparaître avant 20 ans. Par ailleurs, dans une lignée familiale, les symptômes apparaissent de plus en plus tôt au cours des générations. C'est ce qu'on appelle le phénomène d'anticipation. À chaque génération, l'expansion des triplets CAG augmente. Cette expansion est d'autant plus forte qu'elle est transmise par le père. Effectivement, il semblerait que l'instabilité des répétitions CAG survient préférentiellement lors de la spermatogenèse.
Diagnostic
De la même façon que la détection des empreintes génétiques, le diagnostic moléculaire repose sur une analyse de la taille des zones répétées après amplification en PCR. La visualisation des tailles peut se faire soit sur gel soit en analyse de fragments (lire mon article pour comprendre cette technique).
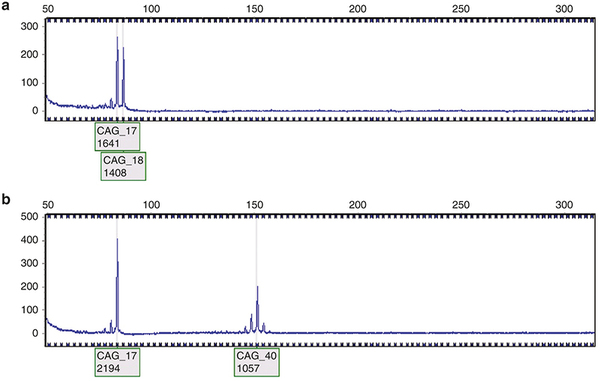
Analyse en fragment chez un sujet sain (a) et un patient atteint de la maladie de Huntigton (b). Le gène du patient (a) présente un allèle à 17 répétitions et un autre à 18 répétitions. Le patient (b) présente un allèle avec une répétition de 40.
Enjeux éthiques
L'absence de traitement fait de cette maladie un cas d'école en éthique médicale. Effectivement, les premiers symptômes peuvent apparaître tardivement et donner le diagnostic ne conduirait à aucune action thérapeutique. C'est donc après discussion entre le patient et une équipe pluridisciplinaire ( Psychologue, généticien, neurologue ...) que le diagnostic pré-symptomatique peut être entrepris. Dans tous les cas, un délai de réflexion est donné au patient.
Le diagnostic prénatal peut également être réalisé au cours de la grossesse en réalisant une amniocentèse. Là aussi la décision est prise à plusieurs lors du CPDPN (Centre Pluridisciplinaire de Diagnostic Pré-Natal) et seulement si les parents sont favorables à une interruption de grossesse. On ne fera jamais de diagnostic chez le fœtus avant d'avoir fait celui des parents. Car si aucun des deux parents n'est porteur de la mutation alors il n'y a aucun risque de transmission au fœtus.
Ces enjeux éthiques sont spécifiques aux maladies génétiques qui se déclarent tardivement. Et lorsque l'on voit que la FDA a autorisé 23andMe (alias google) à dépister les gènes de susceptibilité à la maladie d'Alzheimer, sans passer pas une équipe médicale, on a de quoi se poser des questions.
Un futur traitement ?
Toujours pas de traitement à ce jour. Mais un espoir avec CRISPR-Cas9. Allez jeter un œil sur cet article prometteur : "Permanent inactivation of Huntington's disease mutation by personalized allele-specific CRISPR/Cas9"
Références
Ce site est versionné sur GitHub. Vous pouvez corriger des erreurs en vous rendant à cette adresse
Go Top
comments powered by Disqus