Toutes les cellules de votre corps sont constituées du même génome. Vous obtiendrez toujours le même texte en séquençant l'ADN provenant d'un morceau d'estomac, de cerveau ou de peau (sauf cas très particuliers: mosaïques).
Ce qui fait la différence, c'est l'expression des gènes ou « transcriptome ». C'est-à-dire l'ensemble des ARNs messagers (ARNm) transcrits dans la cellule dont la traduction est responsable du phénotype cellulaire. Par exemple, les cellules de votre rétine expriment d'autres gènes que votre estomac. Leurs transcriptomes sont différents.
Une des méthodes pour évaluer le transcriptome est le séquençage des ARN messager ou RNA-seq.
En résumant rapidement (figure ci-dessous) :
À partir d'un tissu, toutes les cellules sont lysées puis les ARNs messagers sont capturés (en général par leurs queues polyadénylées). Ils sont ensuite convertis en ADN complémentaire (ADNc) par une rétrotranscriptase, amplifiés, puis séquencés. L'étape bio-informatique consiste à aligner les reads sur un génome de référence et faire des normalisations pour évaluer quels gènes sont exprimés. Le nombre d'ARNm séquencés d'un gène correspond à son niveau d'expression ou « abondance ».
Finalement, en analysant différents tissus, on obtient une matrice d'expression (voir tableau ci-dessous).
Pour plus de détails sur l'analyse bio-informatique, je vous invite à jeter un oeil sur l'article de bioinfo-fr traitant de ce sujet.
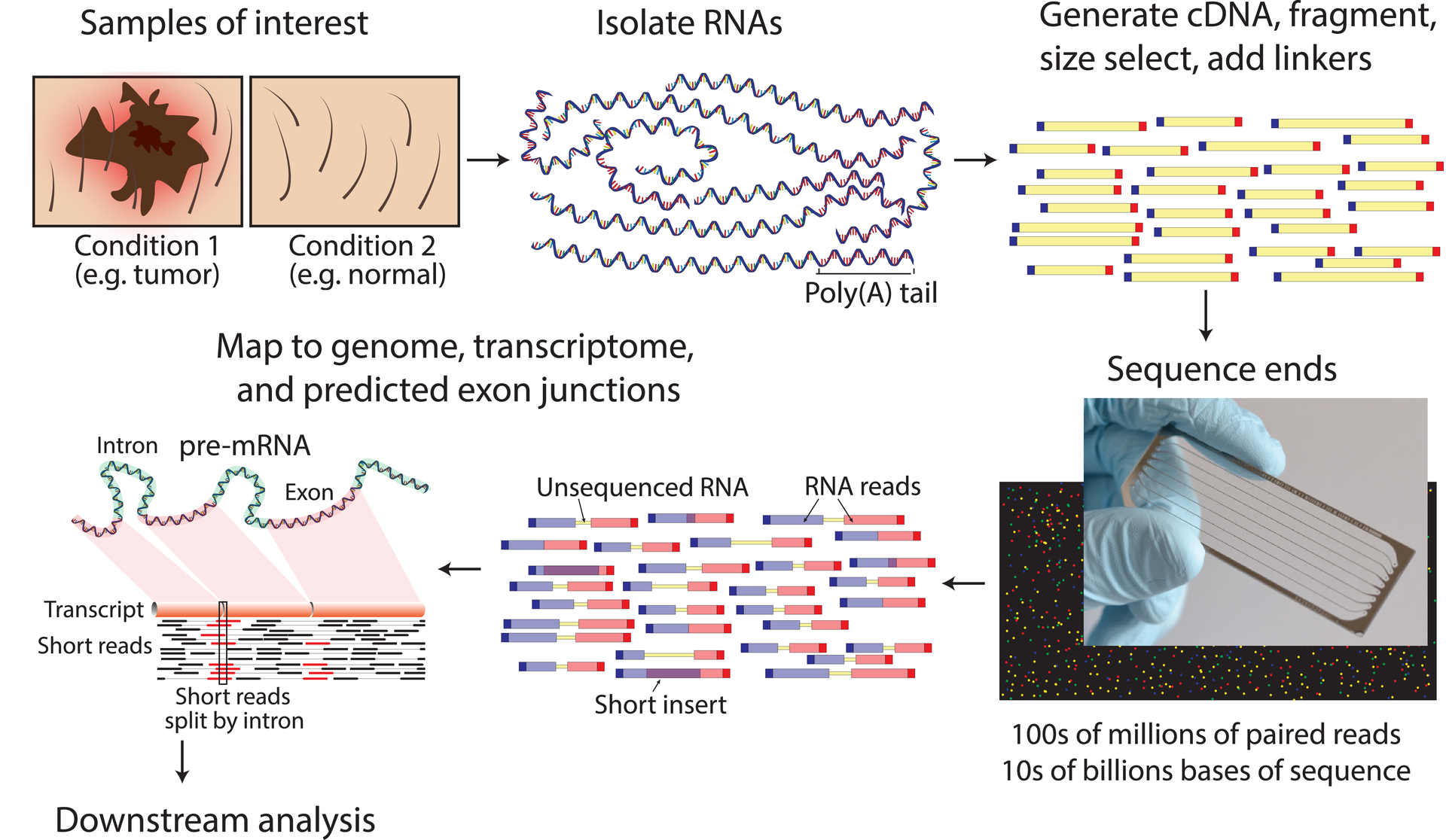
Schéma général de la technologie RNAseq. Dans cet exemple, le séquençage est réalisé sur deux échantillons (tumeur et normal). Les ARNs sont capturés grâce leurs queues polyA, sont convertis en ADNc puis séquencés. Les reads sont alignés sur un génome de référence afin de mesurer l'expression de chaque gène. Cette expression est proportionnelle aux nombres d'ARN s'alignant sur un gène donné Source : Wikipedia

Exemple d'une matrice d'expression comparant deux tissus. Les valeurs du tableau correspondent aux quantités d'ARNm retrouvées par gène et par tissu. L'expression des gènes dans le tissu 1 est différente de celle dans le tissu 2
ScRNA-seq : Nouvelle approche plus résolutive
Le défaut avec la technologie RNA-seq est qu'elle mesure l'expression d'un tissu et pas l'expression d'une cellule. En effet, dans un morceau de cerveau par exemple, il y aura différents types cellulaires (neurone, astrocytes, oligodendrocytes ...) avec des profils d'expression différents. Le RNA-seq vous informe seulement du niveau d'expression de cet ensemble de cellules.
Aujourd'hui, une autre méthode permet de séquencer le transcriptome d'une seule cellule. C'est ce qu'on appelle du Single Cell RNA Seq (ScRNA-Seq).
L'idée est de créer une librairie (Ensemble des fragments d'ADN destinés au séquençage) où chaque ARNm se voit greffer une séquence identifiant sa cellule d'origine (barcode). On peut alors, après séquençage, regrouper les reads entre eux grâce à leurs barcodes et obtenir une matrice d'expression par cellules et par gènes.
Comment étiqueter chaque fragment d'ADN avec sa cellule d'origine ? C'est ce qu'on va voir tout de suite avec la méthode de microfluidique de 10xGenomics.
Isoler les cellules en microfluidique
La microfluidique est une technologie manipulant des fluides dans des microcanaux. Grâce à cette technique, on va pouvoir isoler chaque cellule dans une gouttelette d'huile contenant des réactifs (polymérase, oligonucléotide, retrotranscriptase...) et une bille particulière appelée GEM (Gel bead in EMulsion).
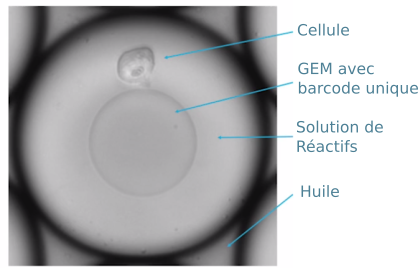
Microgoutelette avec une cellule et une GEM (Gel bead in EMulsion) Source : 10xGenomics
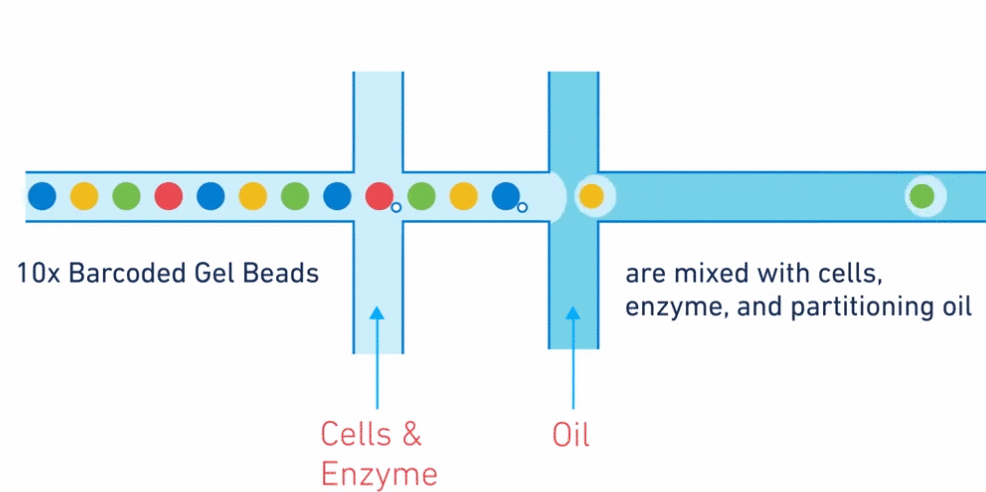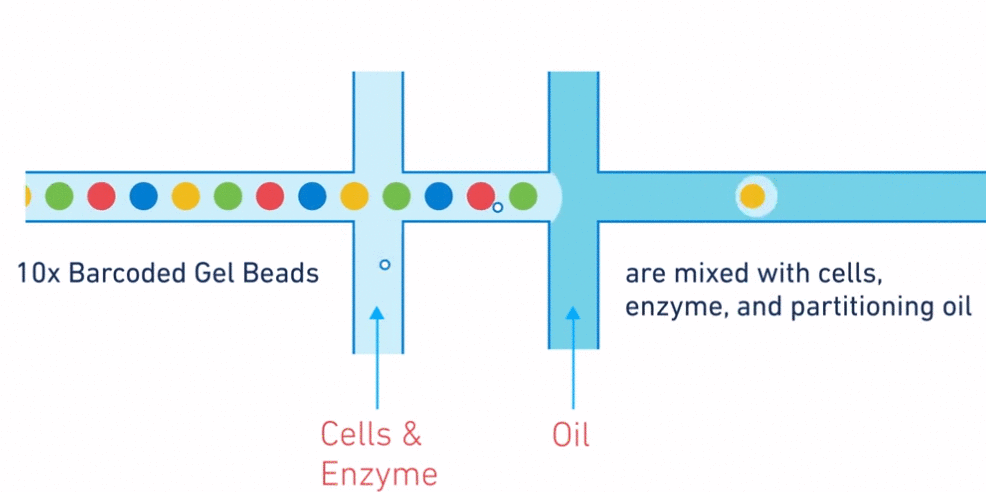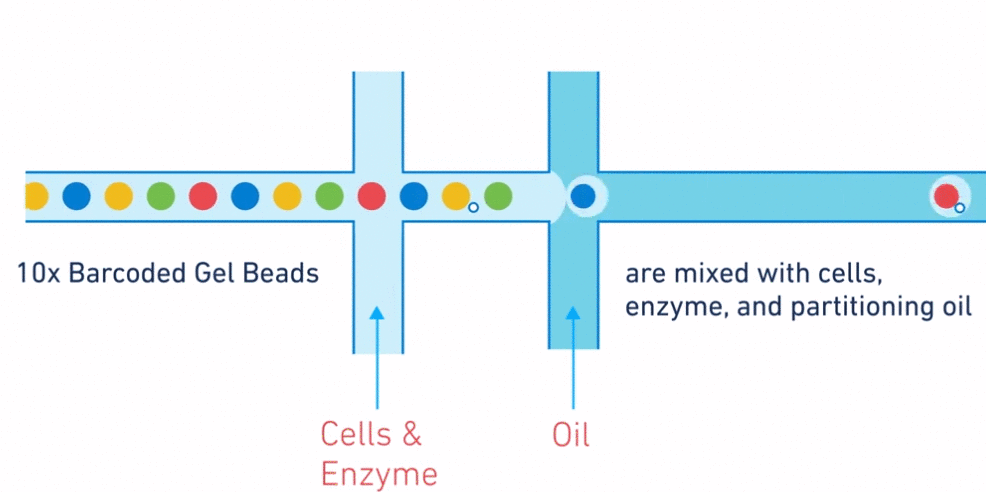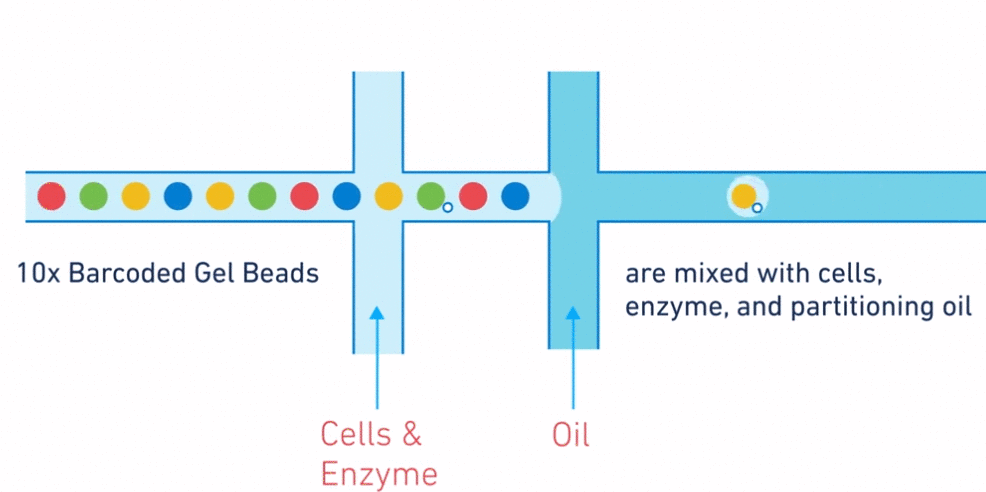
Animation montrant la formation des microgoutelettes en microfluidique. Les GEMs sont définies par un barcode unique représenté ici par une couleur Source : 10xGenomics

Vidéo de microfluidique Source : Dolomite Microfluidics
Chaque cellule a son barcode unique
Chaque GEM est recouverte (figure ci-dessous) de séquences adaptatrices uniques contenant un barcode, un UMI et la séquence PolyT .
- Le barcode est l'identifiant unique à la bille, et donc unique à la cellule. 10xGenomics propose 750 000 barcodes environ.
- L'UMI (Unique Molecular Identifiers) est une courte séquence aléatoire unique à chaque fragment entourant la bille. Il y a donc plusieurs UMI par bille. Cet identifiant est utilisé pour éviter les biais d'amplifications. Si une séquence est malencontreusement trop amplifiée dans une goutte, elle sera détectée, car le même UMI sera représenté plusieurs fois.
- La séquence polyT va permettre la fixation des ARNs messagers par complémentarité avec leurs queues polyA.
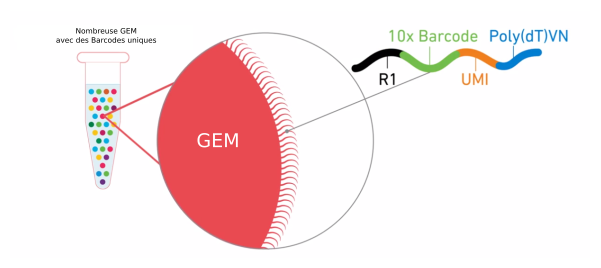
Zoom sur une GEM et les séquences la recouvrant Source : 10xGenomics
La réaction de RNA-seq peut alors se faire dans ce microréacteur. Après lyse de la cellule, les ARNs messagers sont capturés à la surface de la GEM par leurs queues polyA. Et les nouvelles séquences Barcode+UMI+ARNm sont converties en ADNc.
Création d'une librairie et séquençage
Il ne reste plus alors qu'à créer la librairie pour le séquençage. Tous les fragments d'ADNs identifiés par leurs barcodes sont poolés ensemble après avoir enlevé l'huile. Les adaptateurs de séquençage (Illumina) sont ajoutés afin d'obtenir la librairie.
Après le séquençage et l'alignement, il suffira de regrouper les reads provenant d'une même cellule en comparant leurs barcodes pour obtenir une matrice d'expression (tableau ci-dessous).
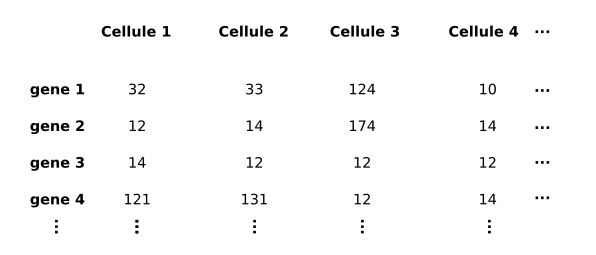
Exemple d'une matrice d'expression en Single Cell RNA Seq. En réalité, il y a des milliers de cellules (autant que de barcode) et au moins 23 000 gènes (pour l'homme). Les valeurs du tableau correspondent à la quantité d'ARNm retrouvé par gène et par cellule
Représentation graphique
On peut alors représenter la matrice d'expression dans un graphique en réalisant une analyse en composantes principales (10x genomics utilise une t-SNE). Chaque point correspond à une cellule. Plus les cellules sont proches sur le graphique, plus leurs expressions génétiques sont similaires.
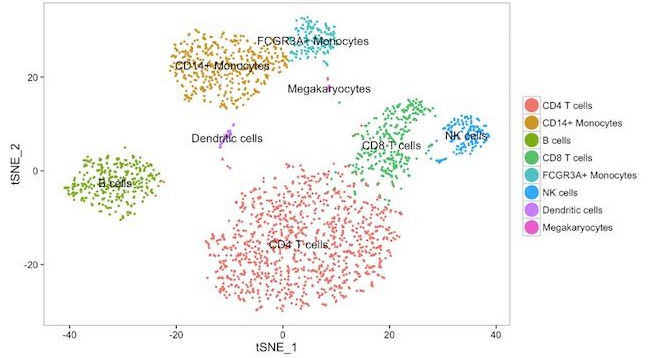
Profil d'expression obtenu à partir des cellules du sang (2,700 cellules mononuclées du sang périphérique PBMC. On visualise après clusterisation les différentes familles. Source : http://satijalab.org/seurat/get_started_v1_4.html
Encore plus parlant, cette vidéo qui montre le profil d'expression des cellules du tissu cérébral dans un repère à 3 axes animé.
What next ?
À l'heure où j'écrivais ce post, je suis tombé sur un article décrivant la technique DropNc-Seq. Une méthode similaire à ce qui vient d'être décrit. Mais au lieu des cellules, ce sont les noyaux qui sont isolés pour le séquençage. On obtient alors le transcriptome nucléaire... Cool hein ?
Références
- RNASeq sur bioinfo-fr
- 10xGenomics
- Dolomite Microfluids
- Massively parallel digital transcriptional profiling of single cells
- Vidéo commerciale
Ce site est versionné sur GitHub. Vous pouvez corriger des erreurs en vous rendant à cette adresse
Go Top
comments powered by Disqus