Aujourd'hui, J'ai eu une soudaine envie de calculer les fréquences des différents types de substitutions dans le génome de James Watson.
A partir d'un fichier contenant des variations par rapport au génome de référence, je me suis amusé à compter le nombre et le type de substitutions nucléotidiques. Et le résultat est loin d'être aléatoire...
Mais d'abord quelques définitions.
Transition et Transversion
On classe les bases azotées ( Adénine,Guanine,Cytosine,Tymine ) en 2 familles: les purines et les pyrimidines.
l'Adénine et la Guanine sont des purines, composées de deux cycles aromatiques.
La Thymine et la Cytosine sont des pyrimidines composées d'un seul cycle.
Gravez-vous en tête que CYtosine et TYmine sont des PYrimidines car ces trois mots contiennent un Y !
Une transition est une substitution entre deux bases sans changement de famille. C'est à dire une purine qui devient une autre purine, ou alors une pyrimidine qui devient une autre pyrimidine.
Une transversion est associée à un changement de famille. C'est une purine qui se transforme en pyrimidine ou l'inverse.
L'image ci-dessous résume tous les cas possibles.
Schéma de toutes les substitutions possibles. En bleu les transitions , en rouge les transversions. Il y a 2 fois plus de transversions que de transitions
Notation consensus
Une substitution s'écrit souvent sous la forme X>Y et se lit X donne Y. Par exemple la substitution A>T signifie que l'allèle de référence est un A tandis que l'allèle alternative est un T.
Si nous résumons tous les cas possibles, il y a au total :
- 4 transitions : A>G, G>A, C>T, T>C
- 8 transversions : A>C,C>A,G>T,T>G,G>C,C>G,A>T,T>A
Mais rappelons-nous que l'ADN est double brin avec une complémentarité des bases. L'Adénine est toujours en face d'une Tymine et la Guanine toujours en face d'une Cytosine. Si il y a une mutation sur un brin, disons un A>G , alors il y a sur le brin complémentaire un T>C. Ces deux notations sont donc équivalente. On utilisera toujours par la suite la notation ou la base de référence est soit une Tymine soit une Cytosine. (T>C au lieu de A>G).
En reprenant notre combinatoire précédente, on se retrouve alors avec :
- 2 transitions : C>T, T>C
- 4 transversions : C>A, T>G, C>G, T>A
Si les mutations sont aléatoires alors nous devons observer 2 fois plus de transversions que de transitions.
Vérifions cela en regardant quelles sont les mutations substitutives qui différencient James Watson au génome de référence hg19.
Allez directement au résultat si Linux vous donne des boutons.
Téléchargement des données nécessaires
Génome de référence: hg19.fa
Le génome humain dans sa version hg19 est disponible sur ucsc.
$ wget http://hgdownload.cse.ucsc.edu/goldenPath/hg19/bigZips/hg19.2bit
Il faut utiliser l'outil twoBitToFa pour convertir le génome au format fasta.
$ wget http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/twoBitToFa
$ chmod +x twoBitToFa
$ ./twoBitTo hg19.2bit hg19.2bit hg19.fa
Substitution de James Watson: pgWatson.txt.gz
Ce fichier contient l'ensemble des variations de James Watson par rapport au génome de référence.
$ wget http://hgdownload.cse.ucsc.edu/goldenpath/hg19/database/pgWatson.txt.gz
Téléchargement de bedtools
bedtools est un outil permettant de manipuler des fichiers au format bed. Ce sont des fichiers contenant des régions génomiques de la forme :
chromosome debut fin
C'est un peu le couteau suisse du bioinformaticien. Si vous ne l'avez toujours pas, Veuillez suivre les instructions pour l'installer ou taper les commandes suivantes.
$ wget https://github.com/arq5x/bedtools2/releases/download/v2.25.0/bedtools-2.25.0.tar.gz
$ tar -zxvf bedtools-2.25.0.tar.gz
$ cd bedtools2
$ make
$ sudo make install
Bash pour Nidja
Récupérer les régions des variants de James Watson
Les trois première colonnes du fichier pgWatson.txt.gz correspondent à la région génomique (chromosome-début-fin) de la substitution. C'est notre fichier bed que nous allons générer avec la commande suivante.
zcat pgWatson.txt.gz|cut -f1,2,3 > region.bed
Récupération des bases de référence
Le fichier region.bed nous permet de récupérer les bases de références depuis hg19. A l'aide de bedtools, créer le fichier ref_bases.txt.
bedtools getfasta -fi hg19.fa -bed region.bed -fo /dev/stdout | awk 'NR%2 == 0 {print $0}' > ref_bases.txt
Récupération des bases alternative
Créez aussi le fichier alt_bases.txt contenant les bases alternatives depuis pgWatson.txt.gz à la colonne 5.
zcat pgWatson.txt.gz|cut -f5|awk -F "/" 'NF==2{print $2} NF==1{print $1}' > alt_bases.txt
Concaténation des deux fichiers
On fusionne ref_bases.txt et alt_bases.txt dans un fichier. Celui ci contient alors une colonne pour la base de référence et une autre pour la base alternative. Les bases sont toutes converties en majuscules.
paste -d '' ref_bases.txt alt_bases.txt|tr '[:lower:]' '[:upper:]' > substitution.txt
Notation consensus
Toutes les substitutions du fichier précédent sont transformées de façon à toujours avoir une Cytosine ou une Tymine en référence.
cat substitution.txt |sed -e 's/AG/TC/' -e 's/GA/CT/' -e 's/AC/TG/' -e 's/GT/CA/' -e 's/GC/CG/' -e 's/AT/TA/' > consensus.txt
Compatage des substitutions
cat consensus.txt|sort|uniq -c
Nous obtenons alors :
697641 CT
683841 TC
183890 CA
173279 TG
172228 CG
146872 TA
466 GG
403 AA
395 CC
369 TT
Il ne reste plus qu'à faire un jolie graphique!
Resultats
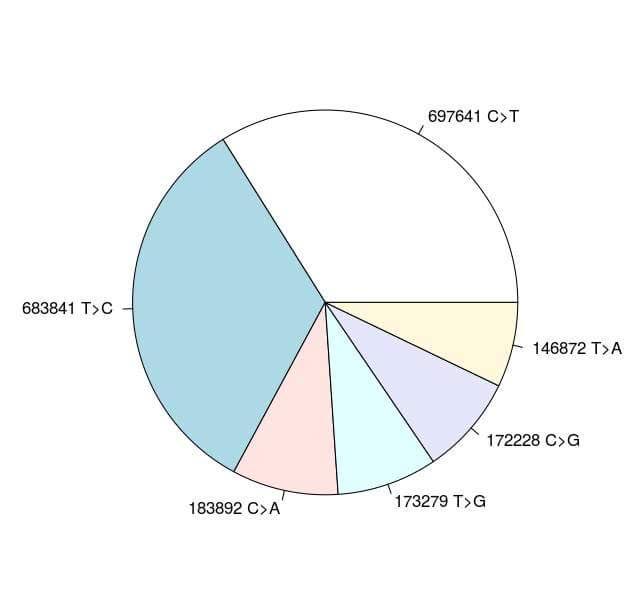
Représentation des différentes substitutions retrouvées dans le génome de James Watson.
Ce graphique représente les différente substitutions retrouvées chez James Watson par rapport au génome de référence. Et comme vous pouvez le constater, cela ne colle absolument pas avec notre hypothèse des mutations aléatoires. Les 2 transitions possibles représentent deux tiers des substitutions, la où les 4 transversions ne représentent qu'un tiers. Il y a donc 2 fois plus de transitions que de transversions.
Ce phénomène est bien connu et s'explique par le fait qu'une transition est plus facile chimiquement à réaliser qu'une transversion. Les structures chimiques étant semblable.
D'après la littérature, ce rapport passe de 2 à 3 dans les régions codantes. En effet une transversion est plus susceptible de modifier l'acide aminé et se retrouve éliminée par la sélection naturelle.
La connaissance de ce rapport est utilisé dans les modèles d’évolution moléculaire comme le modèle de Kimura.
Il s'agit aussi d'un indicateur de qualité de séquençage haut débit. Si le rapport n'est pas retrouvé, il y a fort à parier que des erreurs de séquences se sont produites.
Voilà ! Si vous avez d'autres explications je suis preneur !
Références
- Center for statistical genetics
- Biostar
- Effective filtering strategies to improve data quality from population-based whole exome sequencing studies
Ce site est versionné sur GitHub. Vous pouvez corriger des erreurs en vous rendant à cette adresse
Go Top
comments powered by Disqus